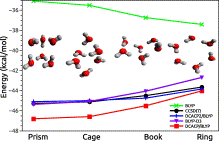
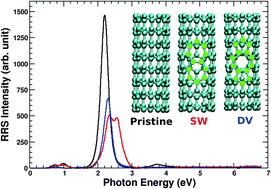
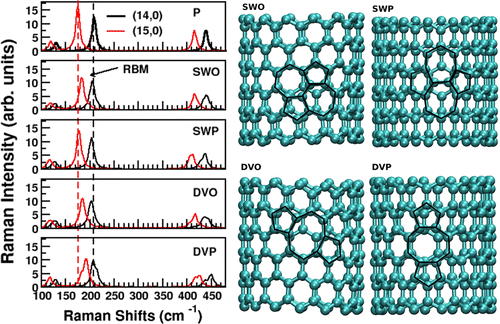
Author
Weibin Chu, Wissam A. Saidi and Oleg Prezhdo
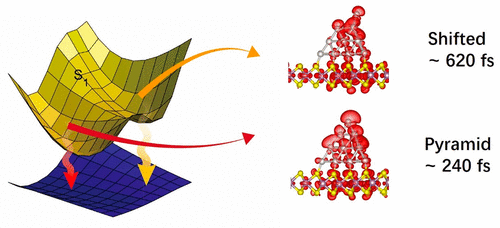
Multiple experiments provide evidence for photovoltaic, catalytic, optoelectronic, and
plasmonic processes involving hot, i.e., high energy, electrons in nanoscale materials.
However, the mechanisms of such processes remain elusive, because electrons rapidly lose
energy by relaxation through dense manifolds of states. We demonstrate a long-lived hot
electron state in a Pt nanocluster adsorbed on the MoS2 substrate. For this purpose, we
develop a simulation technique, combining classical molecular dynamics based on machine
learning potentials with ab initio nonadiabatic molecular dynamics and real-time
time-dependent density functional theory. Choosing Pt20/MoS2 as a prototypical system,
we find frequent shifting of a top atom in the Pt particle occurring on a 50 ps time
scale. The distortion breaks particle symmetry and creates unsaturated chemical bonds.
The lifetime of the localized state associated with the broken bonds is enhanced by a
factor of 3. Hot electrons aggregate near the shifted atom and form a catalytic reaction
center. Our findings prove that distortion of even a single atom can have important
implications for nanoscale catalysis and plasmonics and provide insights for utilizing
machine learning potentials to accelerate ab initio investigations of excited state
dynamics in condensed matter systems.
Author
Yuning Wu, Jeffrey K Wuenschell, Robert Fryer, Wissam A. Saidi, Paul Ohodnicki,
Benjamin Chorpening and Yuhua Duan
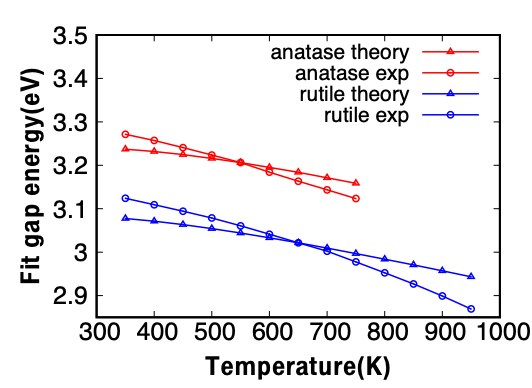
To gain fundamental understanding of the high-temperature optical gas-sensing and
light-energy conversion materials, we comparatively investigate the temperature effects
on the band gap and optical properties of rutile and anatase TiO2 experimentally and
theoretically. Given that the electronic structures of rutile and anatase are
fundamentally different, i.e. direct band gap in rutile and indirect gap in anatase, it
is not clear whether these materials exhibit different electronic structure
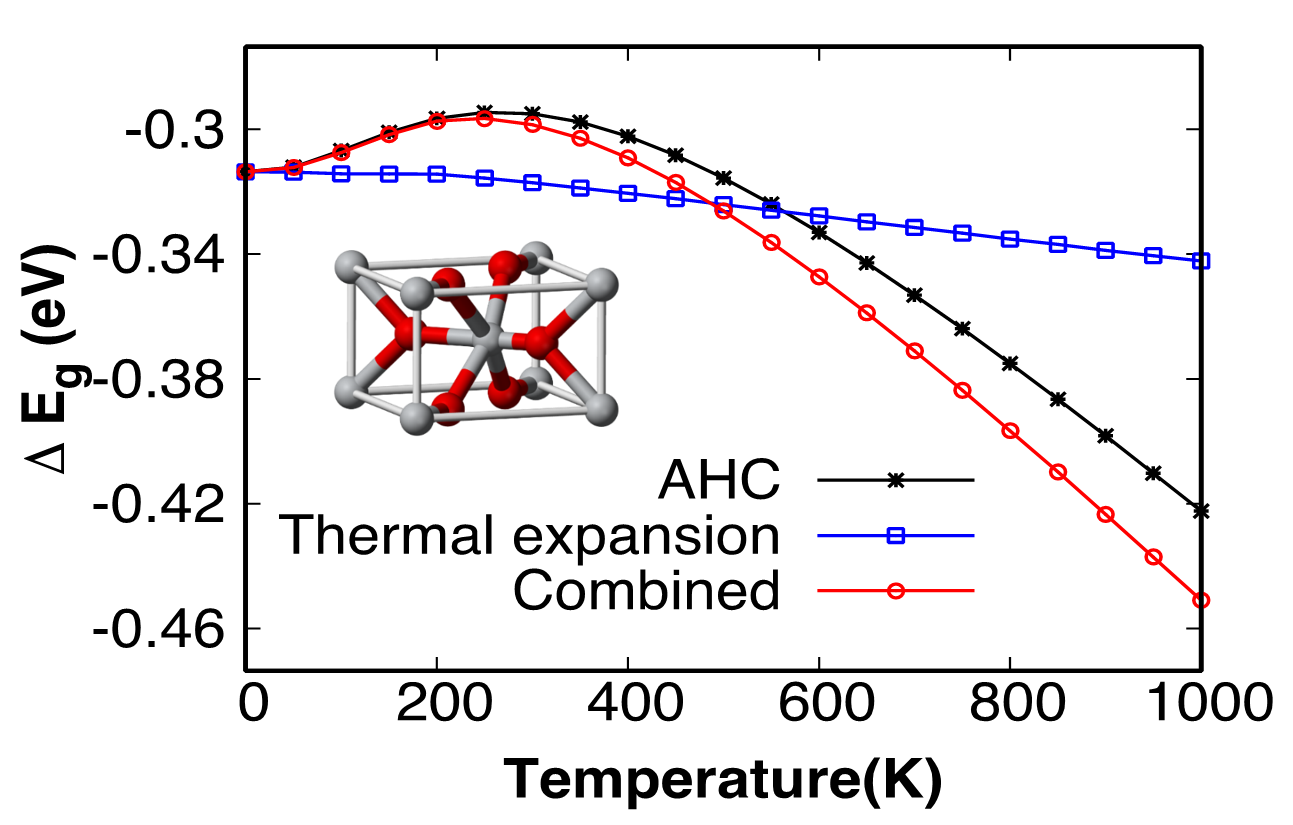
renormalizations with temperature. Using ab initio methods, we show that the
electron-phonon interaction is the dominant factor for temperature band gap
renormalization compared to the thermal expansion. As a result of different
contributions from the acoustic and optical phonons, the band gap is found to widen with
temperature up to 300 K, and to narrow at higher temperatures. Our calculations suggest
that the band gap is narrowed by about 147 meV and 128 meV at 1000 K for rutile and
anatase, respectively. Experimentally, for rutile and anatase TiO2 thin films we
conducted UV-Vis transmission measurements at different temperatures, and analyzed band
gaps form the Tauc plots. For both TiO2 phases, the band gap is found to decrease for
temperature above 300 K quantitatively, agreeing with our theoretical results. The
temperature effects on the dielectric functions, the refractive index, the extinction
coefficient as well as the optical conductivity are also investigated. Rutile and
anatase show generally similar optical properties, but differences exist in the long
wavelength regime above 600 nm, where we found that the dielectric function of rutile
decreases while that of anatase increases with temperature increase.
Author
Weibin Chu, Qijing Zheng, Oleg V. Prezhdo, Jin Zhao and Wissam A. Saidi
Recently we proposed that defect tolerance in the hybrid perovskites is due to their
characteristic low-frequency lattice phonon modes that decrease the non-adiabatic
coupling and weaken the overlap between the free carrier and defect states [Sci. Adv. 6
7, eaaw7453 (2020)]. Kim and Walsh disagree with the interpretation and argue that there
are flaws in our employed methodology. Herein we address their concerns and show that
their conclusions are not valid due to misunderstandings of nonadiabatic transitions.
Author
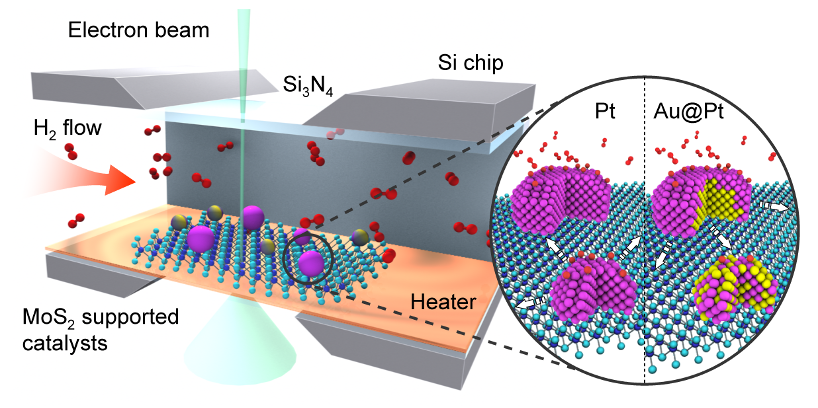
Boao Song, Timothy T. Yang, Yifei Yuan, Soroosh Sharifi-Asl, Meng Cheng, Wissam A.
Saidi, Yuzi Liu, and Reza Shahbazian-Yassar
The decoration of two-dimensional (2D) substrates with nanoparticles serve as
heterostructures for various catalysis applications. Deep understanding of catalyst
degradation mechanisms during service conditions is crucial to improve the catalyst
durability. Herein, we studied the sintering behavior of Pt and bimetallic Au-core
Pt-shell (Au@Pt core-shell) nanoparticles (NPs) on MoS2 supports at high temperatures
under vacuum, nitrogen (N2), hydrogen (H2), and air environments by in situ gas-cell
transmission electron microscopy (TEM). The key observations are summarized as: Effect
of environment: while particle migration and coalescence (PMC) was the main mechanism
that led to Pt and Au@Pt NPs degradation under vacuum, N2 and H2 environments, the
degradation of MoS2 substrate was prominent under exposure to air at high temperatures.
Pt NPs were less stable in H2 environment when compared with the Pt NPs under vacuum or
N2, due to Pt-H interactions that weakened the adhesion of Pt on MoS2. Effect of
nanoparticle composition: under H2, the stability of Au@Pt NPs was higher in comparison
to Pt NPs. This is because H2 promotes the alloying of Pt-Au, thus reducing the number
of Pt at the surface (reducing H2 interactions) and increasing Pt atoms in contact with
MoS2. Effect of nanoparticle size: The alloying effect promoted by H2 was more
pronounced in small size Au@Pt NPs resulting in their higher sintering resistance in
comparison to large size Au@Pt NPs and similar size Pt NPs. The present work provides
key insights into the parameters affecting the catalyst degradation mechanisms on 2D
supports.
Author
Henry O. Ayoola, Cecile S. Bonifacio, Qing Zhu, Cheng-Han Li, Stephen D. House,
Joshua J. Kas, Joerg Jinschek, John J. Rehr. Wissam A. Saidi, and Judith C. Yang
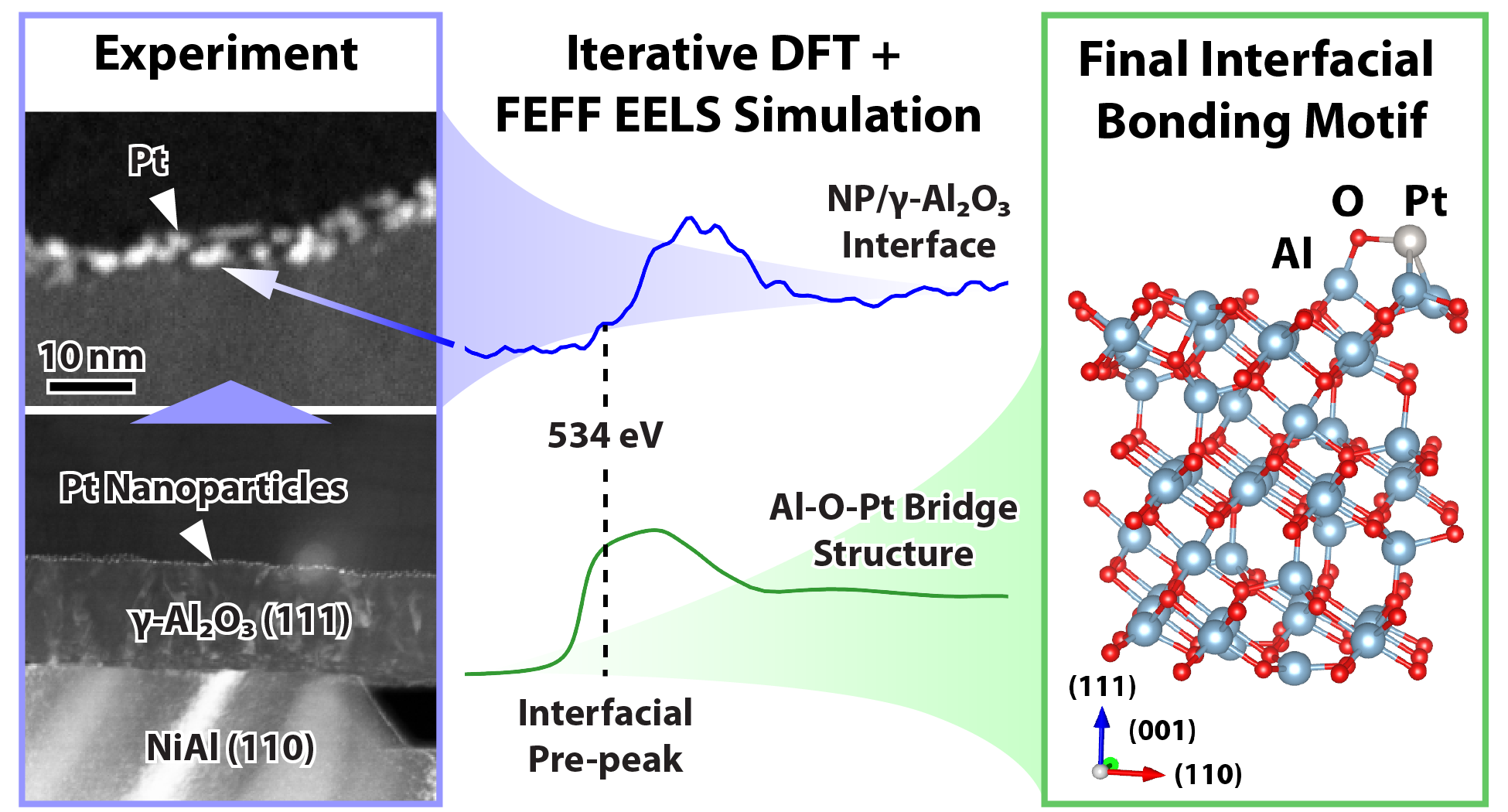
Metal-support interactions significantly affect the performance of heterogeneous
catalysts, of which Pt supported on γ-Al2O3 is one of the most widely used.
Characterizing the bonding of Pt on the γ-Al2O3 surface is key to fully understanding
the catalyst-support interaction. Herein aberration-corrected and monochromated scanning
transmission electron microscopy-based electron energy-loss spectroscopy (STEM-EELS)
were employed on a model Pt/γ-Al2O3(111) catalyst to locally investigate the bonding
between Pt and the γ-Al2O3 support. Differences in the aluminum L2,3-edge and oxygen
K-edge EELS near-edge fine structure between spectra acquired at the Pt/γ-Al2O3(111)
interface and the bulk γ-Al2O3 served as signatures of the interfacial bonding
environment. Fine structure in the interface-localized Al-L2,3 edge suggested a larger
fraction of tetrahedrally coordinated Al atoms at the Pt/γ-Al2O3(111) interface, which
was confirmed by DFT simulations. The interface-localized O-K edge EELS revealed a
pre-peak associated with several types of oxygen bonding. To determine the specific
interfacial O bonding, O-K edge EELS spectra were simulated from an array of
Pt/γ-Al2O3(111) bonding configuration models. The simulated EELS from the interfacial
bonding models revealed an O bonding motif consistent with the experimental O-K edge
EELS fine structure.
Author
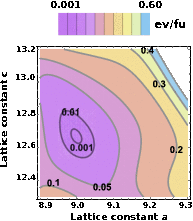
Wissam A. Saidi, Waseem Shadid, and Ivano E. Castelli
The development of statistical tools based on machine learning (ML) and deep networks is
actively sought for materials design
problems. While structure-property relationships can be accurately determined using
quantum mechanical methods, these first-
principles calculations are computationally demanding, limiting their use in screening a
large set of candidate structures. Herein, we
use convolutional neural networks to develop a predictive model for the electronic
properties of metal halide perovskites (MHPs)
that have a billions-range materials design space. We show that a well-designed
hierarchical ML approach has a higher fidelity in
predicting properties of the MHPs compared to straight-forward methods. In this
architecture, each neural network element has a
designated role in the estimation process from predicting complex features of the
perovskites such as lattice constant and
octahedral till angle to narrowing down possible ranges for the values of interest.
Using the hierarchical ML scheme, the obtained
root-mean-square errors for the lattice constants, octahedral angle and bandgap for the
MHPs are 0.01 Å, 5°, and 0.02 eV,
respectively. Our study underscores the importance of a careful network design and a
hierarchical approach to alleviate issues
associated with imbalanced dataset distributions, which is invariably common in
materials datasets.
Author
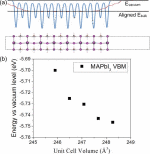
Yongliang Shi, Oleg V. Prezhdo, Jin Zhao, and Wissam A. Saidi
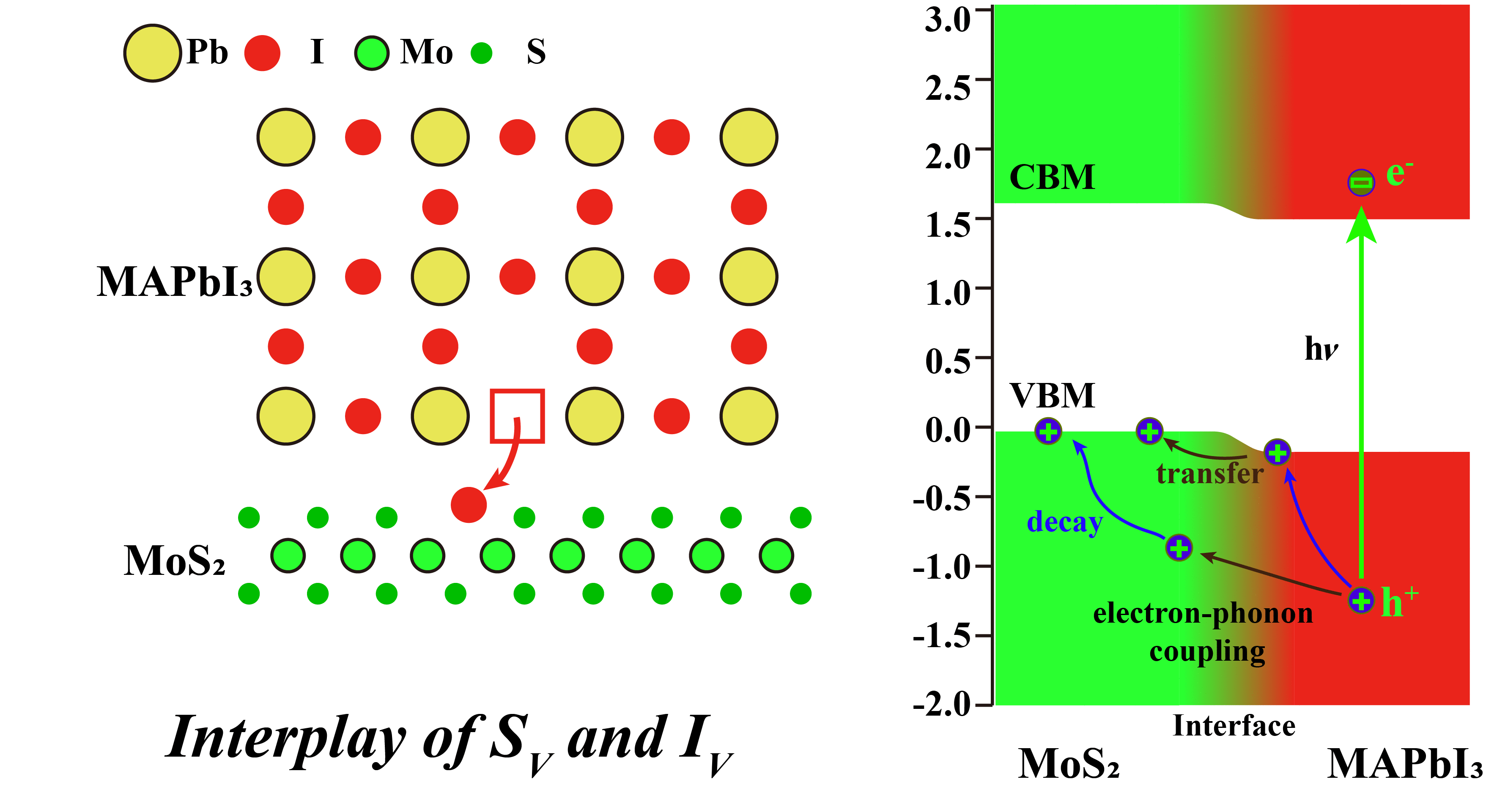
It is crucial to optimize hole transport materials (HTMs) to improve the performance of
metal halide perovskites solar cells. While atomically thin two-dimensional transition
metal chalcogenides (TMDs) are promising HTM candidates due to their high charge
mobility, the nature of the formed type-I heterojunction hampers the transfer of
photo-excited holes. We show that a small concentration of sulfur vacancies (SV) is
already sufficient to stabilize iodine vacancies (IV) at the MAPbI3/MoS2 interface
(SV-to-IV process), to induce an interface dipole moment and to reverse the offset of
the valence band maxima, thus leading to ultrafast hole transport from the absorber to
the electrode. The 0.2-0.8 ps time scale computed from non-adiabatic density functional
theory is in excellent agreement with experiment. Our results prove that the “SV-to-IV”
interface vacancy engineering plays the crucial role in improving the HTM performance of
TMDs.
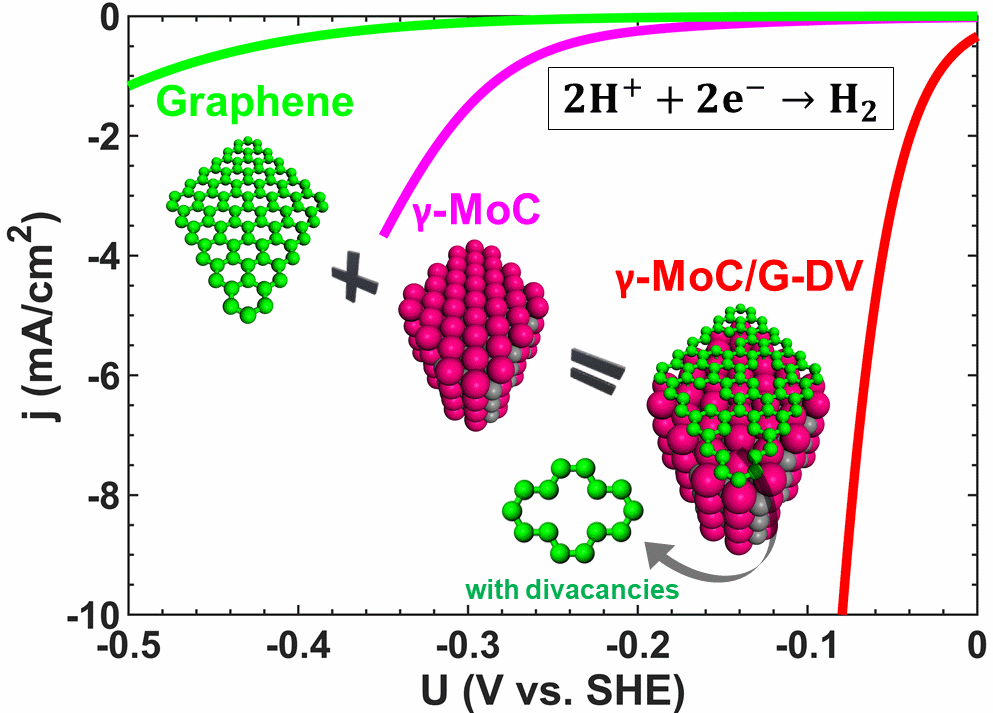
Molybdenum carbides (MoxC) have shown high catalytic activities towards hydrogen
evolution reaction (HER) when coupled with graphene. Herein, we use density functional
theory (DFT) calculations in conjunction with ab initio thermodynamics and
electrochemical modeling on γ-MoC supported graphene to determine the origin of the
enhanced HER activities. In addition to previous claims that graphene’s main role is to
prevent agglomeration of MoxC nanoparticles, we show that the interplay between γ-MoC
coupling and graphene defect chemistry activates graphene for HER. For all γ-MoC
supported graphene systems, the HER mechanism follows Volmer-Heyrovsky pathway with the
Heyrovsky reaction as the rate-determining step. To simulate the electrochemical linear
sweep voltammetry at the device level, we develop a computational current model purely
from the thermodynamic and kinetics descriptors obtained using DFT. This model shows
that γ-MoC supported graphene with divacancies is optimum for HER with an exchange
current density ~1 × 10-4 A/cm2 and Tafel slope ~50 mV/dec-1, which is in good agreement
with experimental results.
Author
Yuning Wu, Wissam A. Saidi, Jeffrey Wuenschell, Terumasa Tadano, Paul R Ohodnicki,
Benjamin Chorpening, and Yuhua Duan
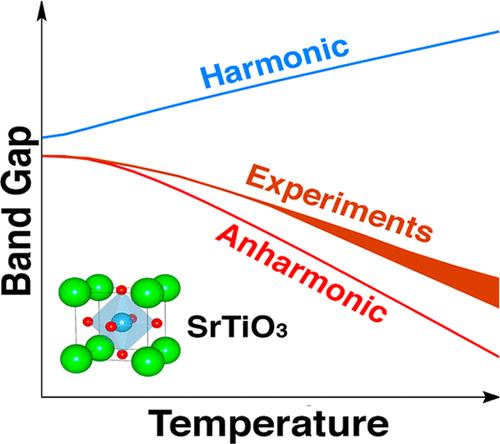
Soft phonon modes in strongly anharmonic crystals are often neglected in calculations of
phonon-related properties. Herein, we experimentally measure the temperature effects on
the band gap of cubic SrTiO3, and compare with first-principles calculations by
accounting for electron–phonon coupling using harmonic and anharmonic phonon modes. The
harmonic phonon modes show an increase in the band gap with temperature using either
Allen–Heine–Cardona theory or finite-displacement approach, and with semilocal or hybrid
exchange-correlation functionals. This finding is in contrast with experimental results
that show a decrease in the band gap with temperature. We show that the disagreement can
be rectified by using anharmonic phonon modes that modify the contributions not only
from the significantly corrected soft modes, but also from the modes that show little
correction in frequencies. Our results confirm the importance of soft-phonon modes that
are often neglected in the computation of phonon-related properties and particularly in
electron–phonon coupling.
Author
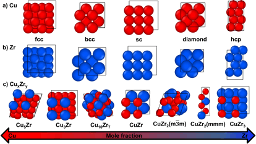
Christopher M. Andolina, Philip Williamson, and Wissam A. Saidi
We show that a deep-learning neural network potential (DP) based on density
functional theory (DFT) calculations can well describe Cu-Zr materials, an
example of a binary alloy system that can coexist in several ordered
intermetallics and as an amorphous phase. The complex phase diagram for Cu-Zr
makes it a challenging system for traditional atomistic force-fields that fail
to describe well the different properties and phases. Instead, we show that a
DP approach using a large database with ~300k configurations can render results
generally on par with DFT. The training set includes configurations of pristine
and bulk elementary metals and intermetallics in the liquid and solid phases in
addition to slab and amorphous configurations. The DP model was validated by
comparing bulk properties such as lattice constants, elastic constants, bulk
moduli, phonon spectra, surface energies to DFT values for identical
structures. Further, we contrast the DP results with values obtained using
well-established two embedded atom method potentials. Overall, our DP potential
provides near DFT accuracy for the different Cu-Zr phases but with a fraction
of its computational cost, thus enabling accurate computations of realistic
atomistic models especially for the amorphous phase.
Author
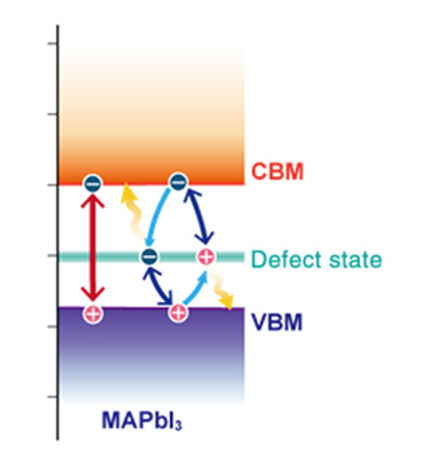
Weibin Chu, Qijing Zheng, Oleg V. Prezhdo, Jin Zhao and Wissam A. Saidi
Low-cost solution-based synthesis of metal halide perovskites (MHPs) invariably
introduces defects in the system, which could form Shockley-Read-Hall (SRH)
electron-hole recombination centers detrimental to solar conversion efficiency. Here, we
investigate the nonradiative recombination processes due to native point defects in
methylammonium lead halide (MAPbI3) perovskites using ab initio nonadiabatic molecular
dynamics within surface-hopping framework. Regardless of whether the defects introduce a
shallow or deep band state, we find that charge recombination in MAPbI3 is not enhanced,
contrary to predictions from SRH theory. We demonstrate that this strong tolerance
against defects, and hence the breakdown of SRH, arises because the photogenerated
carriers are only coupled with low-frequency phonons and electron and hole states
overlap weakly. Both factors appreciably decrease the nonadiabatic coupling. We argue
that the soft nature of the inorganic lattice with small bulk modulus is key for defect
tolerance, and hence, the findings are general to other MHPs.
Author
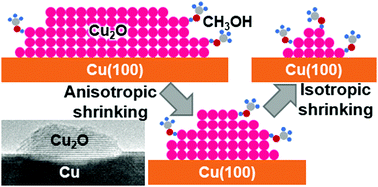
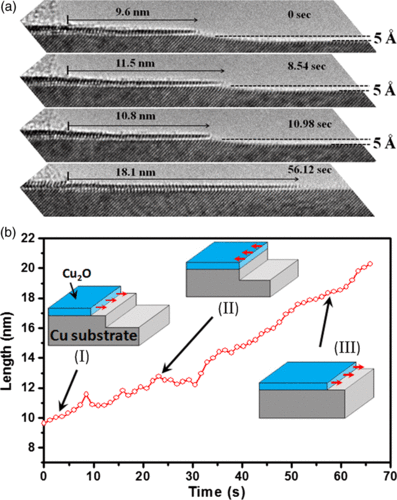
Hao Chi, Matthew T. Curnan, Meng Li, Christopher Andolina, Wissam A. Saidi, Gotz
Veser, and Judith Yang
The structural dynamics of Cu catalyst regeneration from Cu2O under methanol is poorly
understood. In situ Environmental TEM on Cu(100)-supported Cu2O islands reveals a
transition from anisotropic to isotropic shrinking during reduction. Two-stage reduction
is statistically supported and explained by preferential methanol reactivity on Cu2O
nano-islands with DFT simulations.
Author
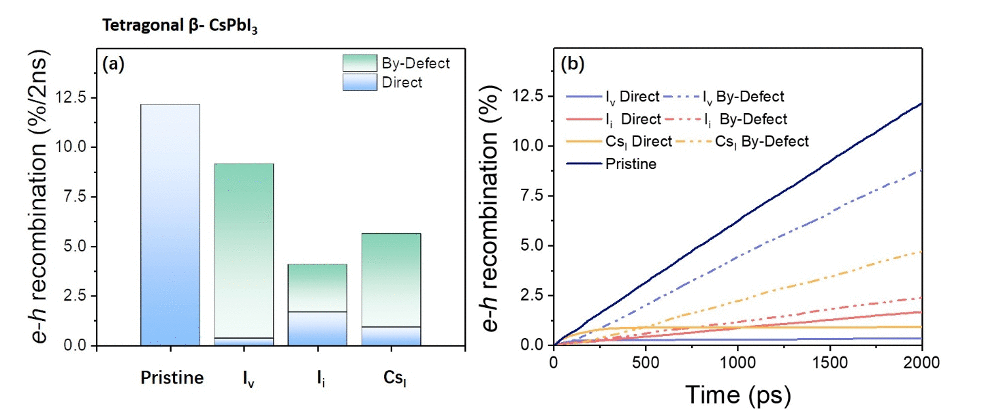
Weibin Chu, Wissam A. Saidi, Jin Zhao and Oleg Prezhdo
Although all‐inorganic lead halide perovskite solar cells have shown tremendous
improvement over the past few years, they are still inferior to the hybrid
organic‐inorganic perovskites in the solar power conversion efficiency. Recently, a
conceptually new β‐CsPbI 3 perovskite has demonstrated an impressive 18.4% efficiency
combined with good thermodynamic stability at ambient conditions. We use ab initio
non‐adiabatic molecular dynamics to show that native point defects in β‐CsPbI 3 are
generally benign for non‐radiative charge recombination, regardless of whether they
introduce shallow or deep trap states. Moreover, formation of new covalently bound
species in the presence of defects slows down the recombination. These results indicate
that halide perovskites do not follow the simple models used to explain defect‐mediated
charge recombination in the conventional semiconductors. The strong tolerance of
electron‐hole recombination against defects arises due to the softness of the perovskite
lattice, which permits separation of electrons and holes upon defect formation, and
allows only low‐frequency vibrations to couple to the electronic subsystem. Both factors
decrease significantly the non‐adiabatic coupling and slow down the dissipation of
electronic energy to heat. We suggest that a halide‐rich synthesis environment may
further improve the efficiency, and propose that strong defect tolerance is general to
metal halide perovskites because they exhibit much lower bulk moduli compared to the
conventional semiconductors used in photovoltaic, photocatalytic, electrocatalytic,
lasing, light‐emitting, detecting and other opto‐electronic devices.
Author
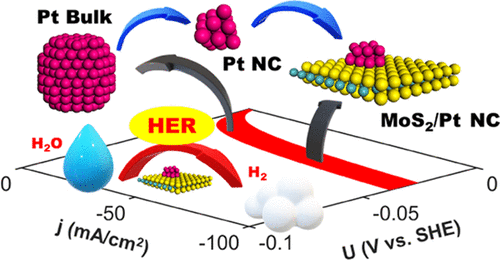
Timothy T. Yang, Teck Leong Tan, and Wissam A. Saidi
The design of efficient and cost-effective platinum-based catalysts for the hydrogen
evolution reaction (HER) is critical for energy sustainability. Herein, we report high
catalytic activity toward HER on the edges of platinum nanoclusters (NCs) supported on
single-layer molybdenum disulfide and provide a direct link between ab initio
calculations and electrochemical experiments. We determine the active catalytic sites
using a cluster expansion method in conjunction with an ab initio thermodynamic approach
and show that the system is thermodynamically active at HER reversible potential under
electrochemical conditions. We also show that the preferred HER mechanism is the
Volmer–Tafel pathway with the Volmer reaction as the rate-determining step. Using a
Butler–Volmer kinetic model to simulate a linear sweep voltammogram, we obtain an
exchange current density of 10–3–10–2 A/cm2, which is in the same order as those
measured for Pt(111) and supported Pt NCs. Importantly, we show that, contrary to
expectations, the enhanced HER mechanism is only attributable to the edges of the
supported Pt NCs but not due to metal–support interactions. Our findings are general and
applicable to NCs with different sizes and shapes on various supports as well as to
different catalytic reactions.
Author
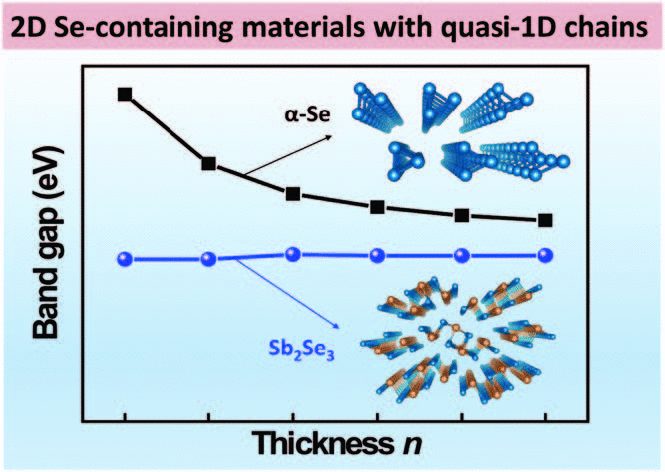
Yawen Li, Yuanhui Sun, Guangren Na, Wissam A. Saidi and Lijun Zhang
The two-dimensional (2D) atomically thin layered materials have attracted significant
attention for constructing next-generation integrated electronic and optoelectronic
devices. A special class of 2D materials composed of quasi one-dimensional (1D) atomic
chains that show intriguing properties are less studied. Here, two Se-containing 2D
layered materials α-Se and Sb2Se3 that have quasi-1D atomic chains are investigated via
first-principles electronic structure calculations. Results shows that the electronic
properties of n-monolayers (n-MLs) stacked α-Se and Sb2Se3 exhibit distinct
layer-dependence electronic properties. The band gap of 2D α-Se remarkably decreases
with increasing thickness, whereas the band gap of 2D Sb2Se3 show negligible change with
thickness. The evolution of lattice phonon frequencies with thickness also show similar
distinction. The underpinnings of the diverse electronic properties are attributed to
the different electronic coupling among the layers of α-Se and Sb2Se3 that results in
different van der Waals interactions among chains/layers. Our study demonstrates the
rich diversity in the properties of 2D layered materials composed of lower-dimensional
structural motifs.
Author
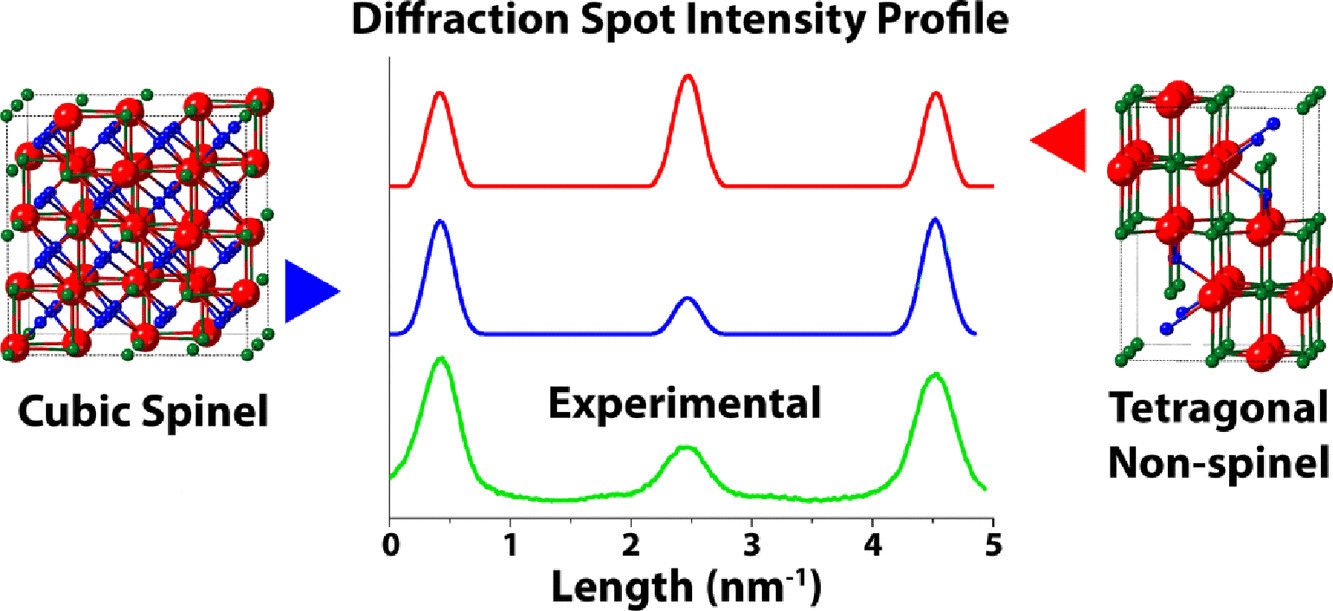
Henry O. Ayoola, Stephen D.House, Cecile S.Bonifacio, Kim Kisslinger, Wissam
A.Saidi, and Judith C.Yang
Single crystal and textured polycrystalline γ-Al2O3 thin films were synthesized by
oxidation of NiAl(110) in air at 850°C for 1 and 2 h, respectively, and used to evaluate
the accuracy of two spinel-based and two nonspinel models by comparison of selected-area
electron diffraction (SAED). The lattice interplanar distances derived from the
polycrystalline SAED pattern most closely matched the cubic spinel γ-Al2O3 model. The
single-crystal SAED spot pattern showed symmetry consistent with both the cubic spinel
and tetragonal nonspinel models, however, the Al cation distribution better matched the
cubic spinel model based on the relative intensities of diffraction spots. Our work
indicates that the traditional cubic spinel model is a more accurate model of γ-Al2O3
than the other models considered. The spinel-based monoclinic model is also more
accurate than the monoclinic nonspinel model. The understanding of the relative accuracy
of the different models is key for simulating γ-Al2O3 containing systems and is of
general interest for the metal oxide and ceramic communities.
Author
Nicholas Smith, Brian Gleeson, Wissam A. Saidi, Anne Kvithyld, and Gabriella Tranell
Al–Mg alloys are known to suffer from problematic oxidation during melting, refining,
and casting. The use of a CO2/air cover gas is known to minimize this oxidation;
however, a mechanistic understanding of the beneficial inhibiting effect is lacking. A
series of thermogravimetric experiments were conducted under a variety of different
CO2-containing atmospheres at 750 °C to elucidate the inhibiting effect.
Characterization of the oxide layer was done by surface and cross-sectional analysis in
the electron microscope and X-ray photoelectron spectrometry (XPS) depth profiling. It
was found that additions of as little as 5% CO2 to air delayed the onset of breakaway
oxidation for at least 7 h and gave a notable reduction in the mass gain compared to
that seen upon exposure to air at 750 °C. The XPS depth profile showed a
carbon-containing layer due to adsorbed CO2 at the top surface of the oxide layer. It
was inferred that this carbon-containing layer slowed the transport of Mg vapor from the
metal through the oxide layer, resulting in a reduction in the amount of Mg vapor
available for oxidation.
Author
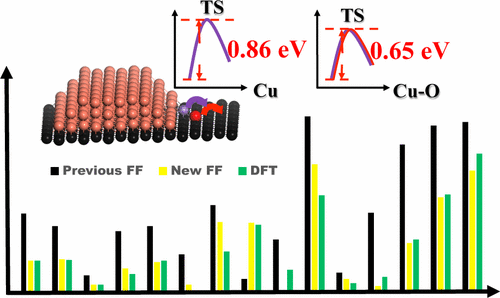
Matthew T. Curnan, Christopher M. Andolina, Meng Li, Qing Zhu, Hao Chi, Wissam A.
Saidi, and Judith C Yang
Current fundamental understanding of the reaction mechanisms controlling Cu oxidation
encompasses early-stage chemisorption and O surface diffusion, as well as later stage Cu
oxide nano-island nucleation and growth. This understanding cannot broadly predict
preferential Cu oxide formation on competing surface defects. Improving understanding on
how to control preferential oxide formation can lead to more effective corrosion
mitigation and Cu/Cu2O catalyst optimization strategies. Computational methods, such as
Density Functional Theory (DFT) and Reactive Force Field (RFF) Molecular Mechanics (MM),
linked by a multiscale approach can calculate early-stage O adsorption and diffusion
energetics on simulated structures comparable to experimental surface defects.
Experimental methods, like Environmental Transmission Electron Microscopy (ETEM), can
characterize later stage preferential Cu oxide formation on competing surface defects.
This study aspires to illustrate consistency between early and later stage oxidation
properties, finding whether computationally modeled differences in O diffusion
energetics can be used to explain experimentally observable oxide formation preferences
along Cu(011) stepped defects. Upon determining which energetics can be applied to
reconcile experimental and computational results, edge-to-edge O diffusion mechanisms
are found to contribute to oxide island formation over edge-to-terrace mechanisms.
Further analysis determines which arrangements of stepped defects can lead to selective
oxidation on competing adjacent stepped defects, reviewing the corners formed by these
defects to characterize experimental outcomes.
Author
Kayla A. Cooley, Rajeh Alsaadi, Ramya L. Gurunathan, Anna C. Domask, Lauren
Kerstetter, Wissam A. Saidi, and Suzanne E. Mohney
The orientation of selected metals (Pd, Ni, Al, and Co) deposited on WSe2 by physical
vapor deposition was examined using transmission electron microscopy and selected area
electron diffraction. We discovered that Ni demonstrates room-temperature epitaxy,
similarly to other face centered cubic (FCC) metals Au, Ag, and Cu. These epitaxial
metals exhibit the following orientation relationship, where M stands for metal: M() ||
WSe2 (); M[] || WSe2 []. Hexagonally close-packed Co, and FCC Pd and Al, were not
epitaxial on deposition; however, Pd became epitaxial after annealing at 673 K for 5 h.
To uncover critical variables for epitaxial growth, we correlated our experimental work
and reports from the literature on Cu, Ag, and Au with density functional theory
calculations of the energetics of metal atoms on the surface of WSe2 and thermodynamic
calculations of metal-W-Se phase equilibria. Furthermore, we compared the findings to
our previous work on metal/MoS2 systems to draw conclusions more generally applicable to
epitaxial growth of metals on transition metal dichalcogenides (TMDs). We observed that
epitaxy of metals on TMDs can occur when there is a match in crystallographic symmetry,
even with a large lattice mismatch, and it is favored by metals exhibiting a low
diffusion barrier on the TMD surface. However, reaction processes between the metal and
WSe2 can prevent epitaxy even when the other factors are favorable, as occurred for
Al/WSe2 with the formation of aluminum selenide, tungsten aluminide, and elemental
tungsten. Consideration of crystallographic symmetry, surface diffusion barriers, and
reactivity can be used to predict room-temperature epitaxy in other metal/TMD systems.
Author
Linfeng Zhang, Jiequn Han, Han Wang, Wissam A. Saidi, Roberto Car and Weinan E
Machine learning models are changing the paradigm of molecular modeling, which is a
fundamental tool for material science, chemistry, and computational biology. Of
particular interest is the inter-atomic potential energy surface (PES). Here we develop
Deep Potential - Smooth Edition (DeepPot-SE), an end-to-end machine learning-based PES
model, which is able to efficiently represent the PES for a wide variety of systems with
the accuracy of ab initio quantum mechanics models. By construction, DeepPot-SE is
extensive and continuously differentiable, scales linearly with system size, and
preserves all the natural symmetries of the system. Further, we show that DeepPot-SE
describes finite and extended systems including organic molecules, metals,
semiconductors, and insulators with high fidelity.
Temperature can have a dramatic effect on the solar efficiency of methylammonium lead
iodide (CH3NH3PbI3) absorbers due to changes in the electronic structure of the system
even within the range of stability of a single phase. Herein, using first-principles
density functional theory, we investigate the electron band structure of the tetragonal
and orthorhombic phases of CH3NH3PbI3 as a function of temperature. The electron–phonon
interactions are computed to all orders using a Monte Carlo approach, which is needed
considering that the second-order Allen–Heine–Cardona theory in electron–phonon coupling
is not adequate. Our results show that the band gap increases with temperature, in
excellent agreement with experimental results. We verified that anharmonic effects are
only important near the tetragonal–cubic phase transition temperature. We also found
that temperature has a significant effect on the effective masses and Rashba coupling.
At room temperature, electron–phonon coupling is found to enhance the band effective
mass by a factor of 2 and to diminish the Rashba coupling by the same factor compared to
T = 0 K values. Our results underscore the significant impact of electron–phonon
coupling on electronic properties of the hybrid perovskites.
Author
Yexin Feng, Yicheng Zhao, Wen-Ke Zhou, Qi Li, Wissam A. Saidi, Qing Zhao, and
Xin-Zheng Li
The organic–inorganic halide perovskites (OIHPs) have shown enormous potential for solar
cells, while problems like the current–voltage hysteresis and the long-term instability
have seriously hindered their applications. Ion migrations are believed to be relevant.
But the atomistic details still remain unclear. Here we study the migrations of ions in
CH3NH3PbI3 (MAPbI3) at varying temperatures (T’s), using combined experimental and
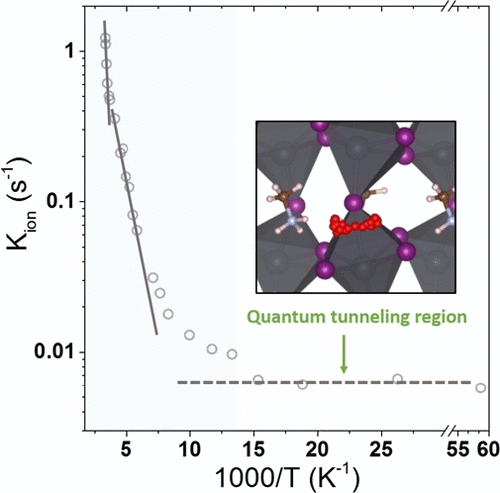
first-principle theoretical methods. Classical hopping of the iodide ions is the main
migration mechanism at moderate T’s. Below ∼270 K, the kinetic constant for ionic
migration still shows an Arrenhius dependency, but the much lower activation energy is
attributed to the migration of H+. A gradual classical-to-quantum transition takes place
between ∼140 and ∼80 K. Below ∼80 K, the kinetic constant becomes T-independent,
suggesting that deep quantum tunneling of H+ takes over. This study gives direct
experimental evidence for the migrations of H+s in MAPbI3 and confirms their quantum
nature.
Author
Yu-Ning Wu, Wissam A. Saidi, Paul Ohodnicki, Benjamin Chorpening, and Yuhua Duan
To gain additional insight into high-temperature functional material properties for
applications in optical gas sensing, the temperature effects on the band gap and optical
properties of rutile TiO2 are investigated using ab initio methods. By analyzing the
contributions from electron-phonon interaction and lattice thermal expansion, we show
that the electron-phonon interaction is the dominant factor for temperature band gap
renormalization. As the temperature increases, the band gap increases until 300 K and
then narrows above 300 K. This behavior results from the acoustic phonons, which widen
the band gap, dominating below 300 K, while the optical phonons, which narrows the band
gap, dominate above 300 K. Our study suggests that the band gap is narrowed by about 138
meV at 1000 K. We also investigated the temperature effects on the dielectric constants,
the refractive index as well as the extinction coefficient. Both the rate of decrease of
the refractive index at 650nm and 800nm as well as the experimentally derived bandgap
agree with experimentally measured data as temperature increases. Our results and
computational methods are of interest for developing high-temperature functional
materials with applications towards gas sensing.
Author
Boao Song, Kun He, Yifei Yuan, Seyyed Sharifi-Asl, Meng Cheng, Jun Lu, Wissam A
Saidi and Reza Shahbazian Yassar
Two-dimensional (2D) substrates decorated with metal nanoparticles offer new
opportunities to achieve high-performance catalytic behavior. However, little is known
on how the substrates control the nucleation and growth processes of the nanoparticles.
This paper presents the visualization of dynamic nucleation and growth processes of gold
nanoparticles on ultrathin MoS2 nanoflakes by in situ liquid-cell transmission electron
microscopy (TEM). The galvanic displacement resulting in Au nuclei formation on MoS2 was
observed in real time inside the liquid cell. We found that the growth mechanism of Au
particles on pristine MoS2 is in between diffusion-limited and reaction-limited,
possibly due to presence of electrochemical Ostwald ripening. A larger size distribution
and more orientation variation is observed for the Au particles along MoS2 edge than on
interior. Differ from pristine MoS2, sulfur vacancies on MoS2 induce Au particle
diffusion and coalescence during growth process. Density functional theory (DFT)
calculations show that the size difference is because the exposed molybdenum atoms at
the edge with dangling bonds can strongly interact with Au atoms, whereas sulfur atoms
on MoS2 interior have no dangling bonds and weakly interact with gold atoms. In
addition, S vacancies on MoS2 generate strong nucleation centers that can promote
diffusion and coalescence of Au nanoparticles. The present work provides key insights on
the role of 2D materials in controlling the size and orientation of noble metal
nanoparticles vital to the design of next generation catalysts.
Author
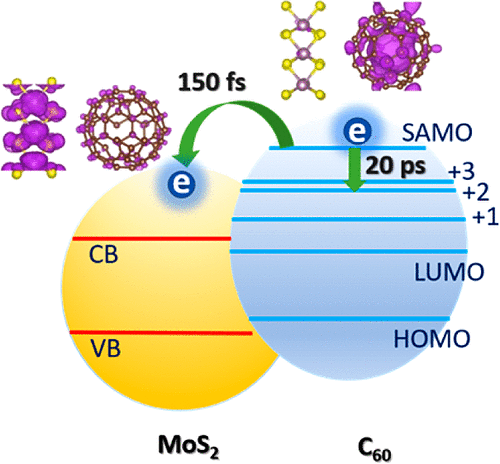
Hongli Guo, Chuanyu Zhao, Qijing Zheng, Zhenggang Lan, Oleg V Prezhdo, Wissam A
Saidi, and Jin Zhao
Hot electron cooling by energy loss to heat through electron–phonon (e–ph) interaction
is an important mechanism that can limit the efficiency of solar energy conversion. To
avoid such energy loss, sufficient charge separation needs to be realized by extracting
hot carriers from the photoconverter before they cool, which requires fast interfacial
charge transfer and slow internal hot carrier relaxation. Using ab initio time-dependent
nonadiabatic molecular dynamics and taking C60/MoS2 as a prototype system, we show that
the superatom molecular orbitals (SAMOs) of fullerenes, which are bound by the central
potential of the whole molecule induced by the charge screening, are ideal media for
charge separation. The diffuse character of SAMOs results in extremely weak e–ph
interaction and therefore acts as a “phonon bottleneck” for hot electron cooling.
Furthermore, it also leads to significant hybridization with other atoms at the
interface that induces fast charge transfer. The interfacial charge-transfer rate at the
C60/MoS2 interface is found to be 2 orders of magnitude faster than the hot electron
cooling from s-SAMO in C60. This conclusion is generally applicable for different carbon
nanostructures that have SAMOs. The proposed SAMO-induced charge separation provides
unique and essential insights into the material design and function for solar energy
conversion.
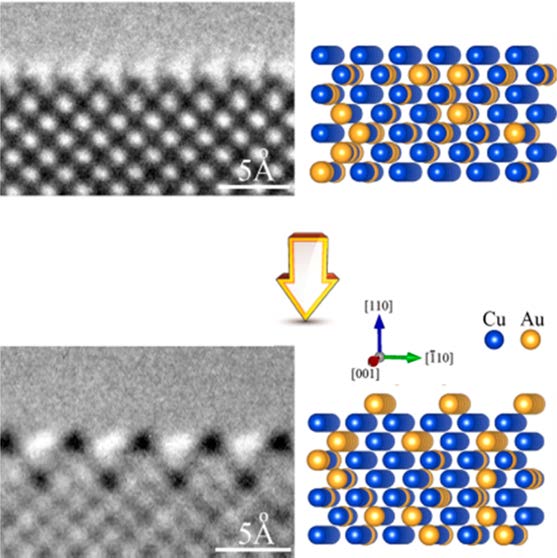
Author
Lianfeng Zou, Wissam A Saidi, Yinkai Lei, Zhenyu Liu, Jonathan Li, Liang Li, Qing
Zhu, Dmitri Zakharov, Eric A Stach, Judith C Yang, Guofeng Wang, and Guangwen Zhou
Using in-situ transmission electron microscopy and atomistic simulations, we report
atomic-scale observations of segregation-induced structure changes in the surface and
subsurface region of a Cu(Au) solid solution in both reductive and oxidative
environments. In a H2 atmosphere, Au segregation induces the formation of a
two-atomic-layer thick ordered surface alloy with an L10 terminated surface layer. By
switching to an O2 atmosphere, the outermost surface develops into an Au-missing row
reconstruction and simultaneously the second layer experiences an order-disorder
transition via intralayer atomic exchanges. The chemical disordering then propagates to
the outermost surface, driven by oxygen-adsorption induced Cu surface segregation. This
transforms the L10 missing-row reconstruction into a non-reconstructed, oxygenated
surface. These observations provide a mechanistic detail regarding the evolution of the
surface and subsurface of this alloy in response to environmental stimuli, and are
relevant to a wide range of technologically relevant processes.
Author
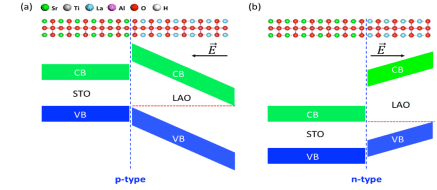
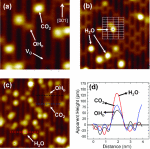
Yanan Wang, Hongli Guo, Qijing Zheng, Wissam A. Saidi and Jin Zhao
Solvated electron states at oxide/aqueous interface represent the lowest energy charge
transfer pathways, thereby playing an important role in photocatalysis and electronic
device applications. However, their energies are usually higher than the conduction band
minimum (CBM), which makes the solvated electrons difficult to utilize in charge
transfer processes. Thus, it is essential to stabilize the energy of the solvated
electron states. In this report, taking LaAlO3/SrTiO3 (LAO/STO) oxide heterostructure
with H2O adsorbed monolayer as a prototypical system, we show using DFT and ab initio
time dependent nonadiabatic molecular dynamics simulation that the energy and dynamics
of solvated electrons can be tuned by the electric field in the polar-nonpolar oxide
heterostructure. Particularly, for LAO/STO with p-type interface, the CBM is contributed
by the solvated electron state when LAO is thicker than 4 unit cells. Furthermore, the
solvated electron band minimum can be partially occupied when LAO is thicker than 8 unit
cells. We propose that the tunability of solvated electron states can be achieved on
polar-nonpolar oxide heterostructure surfaces as well as on ferroelectric oxides, which
is important for charge and proton transfer at oxide/aqueous interfaces.
Author
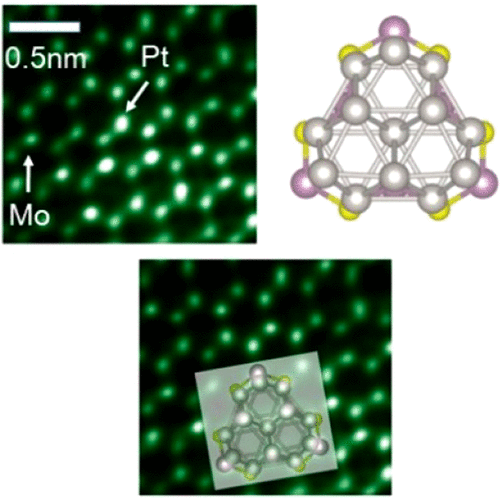
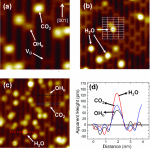
Yongliang Shi, Boao Song, Reza Shahbazian-Yassar, Jin Zhao and Wissam A. Saidi
In nanometer clusters (NCs), each atom counts. It is the specific arrangement of these
atoms that determines the unique size-dependent functionalities of the NCs and hence
their applications. Here, we employ a self-consistent, combined theoretical and
experimental approach to determine atom-by-atom the structures of supported Pt NCs on
MoS2. The atomic structures are predicted using a genetic algorithm utilizing atomistic
force fields and density functional theory, which are then validated using
aberration-corrected scanning transmission electron microscopy. We find that relatively
small clusters grow with (111) orientation such that Pt[11̅0] is parallel to MoS2[100],
which is different from predictions based on lattice-match for thin-film epitaxy. Other
4d and 5d transition metals show similar behavior. The underpinning of this growth mode
is the tendency of the NCs to maximize the metal–sulfur interactions rather than to
minimize lattice strain.
Author
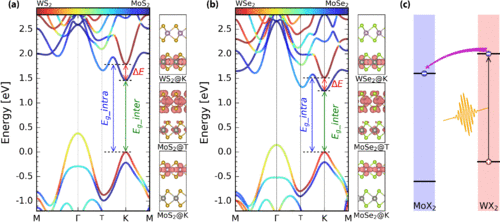
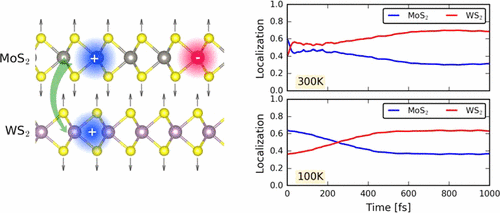
Qijing Zheng, Yu Xie, Zhenggang Lan, Oleg V Prezhdo, Wissam A Saidi, and Jin Zhao
Van der Waals (vdW) heterostructures of transition-metal dichalcogenide (TMD)
semiconductors are central not only for fundamental science, but also for electro- and
optical-device technologies where the interfacial charge transfer is a key factor.
Ultrafast interfacial charge dynamics has been intensively studied, however, the atomic
scale insights into the effects of the electron-phonon (e-p) coupling are still lacking.
In this paper, using time dependent ab initio nonadiabatic molecular dynamics, we study
the ultrafast interfacial charge transfer dynamics of two different TMD heterostructures
MoS2/WS2 and MoSe2/WSe2, which have similar band structures but different phonon
frequencies. We found that MoSe2/WSe2 has softer phonon modes compared to MoS2/WS2, and
thus phonon-coupled charge oscillation can be excited with sufficient phonon excitations
at room temperature. In contrast, for MoS2/WS2, phonon-coupled interlayer charge
oscillations are not easily excitable. Our study provides an atomic level understanding
on how the phonon excitation and e-p coupling affect the interlayer charge transfer
dynamics, which is valuable for both the fundamental understanding of ultrafast dynamics
at vdW hetero-interfaces and the design of novel quasi-two-dimensional devices for
optoelectronic and photovoltaic applications.
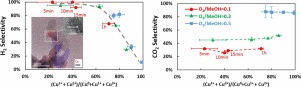
Author
Hao Chi, Christopher M. Andolina, Jonathan Li, Matthew T. Curnan, Wissam A. Saidi,
Guangwen Zhou, Judith C.Yang, and Götz Veser,
Partial oxidation of methanol is a promising reaction for on-board production of high
purity H2 streams for fuel cell applications. In the present work, the influence of Cu
oxidation state on the selectivity of POM catalyzed by Cu/ZnO was investigated via the
use of a microreactor and X-ray photoelectron spectroscopy. A strong correlation between
H2 selectivity and the metallic copper (Cu° ) content of the catalyst was observed,
while, surprisingly, the CO2 selectivity was not significantly affected by the catalyst
oxidation state. Instead, CO2 selectivity showed a strong correlation with O2 partial
pressure, which could be explained by differences in the energy barriers between CO
desorption and CO2 formation from CO* on Cu2O surfaces calculated via first-principles
calculations. Our results indicate that maintaining metallic Cu catalyst during methanol
oxidation could maximize H2 production for use in fuel cells or other clean energy
applications.
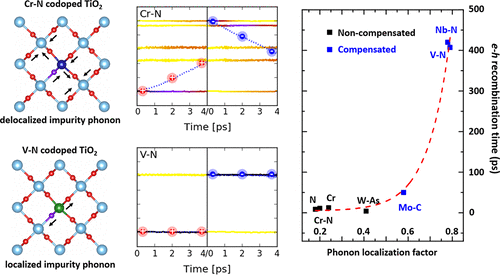
Author
Lili Zhang, Qijing Zheng, Yu Xie, Zhenggang Lan, Oleg Prezhdo, Wissam A. Saidi, and
Jin Zhao
Semiconductor doping is often proposed as an effective route to improving the solar
energy conversion efficiency by engineering the band gap; however, it may also introduce
electron–hole (e–h) recombination centers, where the determining element for e–h
recombination is still unclear. Taking doped TiO2 as a prototype system and by using
time domain ab initio nonadiabatic molecular dynamics, we find that the localization of
impurity-phonon modes (IPMs) is the key parameter to determine the e–h recombination
time scale. Noncompensated charge doping introduces delocalized impurity-phonon modes
that induce ultrafast e–h recombination within several picoseconds. However, the
recombination can be largely suppressed using charge-compensated light-mass dopants due
to the localization of their IPMs. For different doping systems, the e–h recombination
time is shown to depend exponentially on the IPM localization. We propose that the
observation that delocalized IPMs can induce fast e–h recombination is broadly
applicable and can be used in the design and synthesis of functional semiconductors with
optimal dopant control.
Author
Lianfeng Zou, Jonathan Li, Dmitri N Zakharov, Wissam A. Saidi, Eric A. Stach, and
Guangwen Zhou
Using in-situ transmission electron microscopy that spatially and temporally resolves
the evolution of the atomic structure in the surface and subsurface regions, we find
that the surface segregation of Au atoms in a Cu(Au) solid solution results in the
nucleation and growth of a (2×1) missing-row reconstructed, half-unit-cell thick L12
Cu3Au(110) surface alloy. Our in-situ electron microscopy observations and atomistic
simulations demonstrate that the (2×1) reconstruction of the Cu3Au(110) surface alloy
stays as a stable surface structure as a result of the favored Cu-Au diatom
configuration.
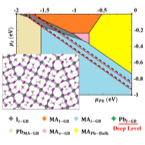
Native point and grain boundary (GB) defects are ubiquitous in methylammonium lead
iodide (MAPbI3) sensitizers employed in solar cells that are polycrystalline in nature.
Here we use density functional theory (DFT) in conjunction with a thermodynamic approach
to determine the stability and electronic properties of all native point defects and
their interplays with Σ5-(210) GB in MAPbI3. The transition levels of charged defects
are investigated with inclusion of electrostatic charge corrections and spin-orbit
coupling. We find that the GB region is a sink for most of the native point defects
under different synthesis conditions. For the crystalline and bi-crystalline MAPbI3 with
Σ5-(210) GB, we find respectively that the p-type antisite defects MAI and PbI, where I
substitutes for MA or Pb, introduce deep levels and both are relatively stable under
I-rich conditions. Hence, I-poor conditions are more preferable for synthesis of MAPbI3
to have defects with electronically benign character.
Author
Qijing Zheng, Wissam A. Saidi, Yu Xie, Zhenggang Lan, Oleg V. Prezhdo, Hrvoje Petek,
and Jin Zhao
Nano Lett., 2017, 17 (10), pp 6435–6442
The van der Waals (vdW) interfaces of two-dimensional (2D) semiconductor are central to
new device concepts and emerging technologies in light-electricity transduction where
the efficient charge separation is a key factor. Contrary to general expectation,
efficient electron–hole separation can occur in vertically stacked transition-metal
dichalcogenide heterostructure bilayers through ultrafast charge transfer between the
neighboring layers despite their weak vdW bonding. In this report, we show by ab initio
nonadiabatic molecular dynamics calculations, that instead of direct tunneling, the
ultrafast interlayer hole transfer is strongly promoted by an adiabatic mechanism
through phonon excitation occurring on 20 fs, which is in good agreement with the
experiment. The atomic level picture of the phonon-assisted ultrafast mechanism revealed
in our study is valuable both for the fundamental understanding of ultrafast charge
carrier dynamics at vdW heterointerfaces as well as for the design of novel quasi-2D
devices for optoelectronic and photovoltaic applications.
Author
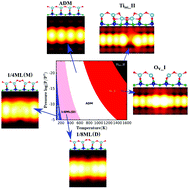
Yongliang shi, Huijuan Sun, Manh Cuong Nguyen, Cai Zhuang Wang, Kai Ming Ho, Wissam
A Saidi and Jin Zhao
Defects on oxide surfaces play a crucial role on the surface reactivity and thus it is
crucial to understand their atomic and electronic structures. The defects on anatase
TiO2(001)-(1×4) surface are found to be highly reactive, however, due to the surface
reconstruction, the defects exhibit complicated characters in different experiments
which make it very challenging to determine their atomic structures. Here we present a
systematic first-principles investigation of the defects on anatase TiO2(001)-(1×4)
surface based on a global-search adaptive genetic algorithm (AGA) and density functional
theory (DFT). For different Ti-O ratios, we identify the low energy defect structures,
investigate their electronic structure using hybrid functional, and map their regions of
stability under realistic conditions. We successfully find novel oxygen vacancy (OV) and
Ti interstitial (Tiini) structures that are different from the conventional ones in
terms of their charge localization, magnetic state, and their
scanning-tunneling-microscopy bright-dark image signature. This provides insight into
the complex geometric and electronic structure of the surface defects, and resolves
several experimental discrepancies.
Author
Yaguang Guo, Wissam A. Saidi, and Qian Wang
Halide perovskites and van der Waals (vdW) heterostructures are both of current interest
owing to their novel properties and potential applications in nano-devices. Here, we
show the great potential of 2D halide perovskite sheets (C4H9NH3)2PbX4 (X = Cl, Br and
I) that were synthesized recently (Dou et al 2015 Science 349 1518–21) as the channel
materials contacting with graphene and other 2D metallic sheets to form van der Waals
heterostructures for field effect transistor (FET). Based on state-of-the-art
theoretical simulations, we show that the intrinsic properties of the 2D halide
perovskites are preserved in the heterojunction, which is different from the
conventional contact with metal surfaces. The 2D halide perovskites form a p-type
Schottky barrier (Φh) contact with graphene, where tunneling barrier exists, and a
negative band bending occurs at the lateral interface. We demonstrate that the Schottky
barrier can be turned from p-type to n-type by doping graphene with nitrogen atoms, and
a low-Φh or an Ohmic contact can be realized by doping graphene with boron atoms or
replacing graphene with other high-work-function 2D metallic sheets such as ZT-MoS2,
ZT-MoSe2 and H-NbS2. This study not only predicts a 2D halide perovskite-based FETs, but
also enhances the understanding of tuning Schottky barrier height in device
applications.
In situ TEM experiments have shown that the oxidation of stepped Cu(100) surface results
in a flat Cu2O film, which is different from the 3D oxide island structure that usually
forms on a flat Cu surface. The mass transport process originating from Cu adatoms that
detach from the step edge is argued to be responsible for the different oxide growth
behavior. Using molecular dynamics in conjunction with a reactive force field (ReaxFF),
we show that the mass transport from the step edge to the flat terrace is enhanced by
the unevenly distributed oxygen adatoms on the step top compared to the flat terrace.
The ReaxFF force field is optimized using density functional theory calculated
energetics and kinetic barriers on various Cu surface models. We investigate two
possible mechanisms that can trigger Cu transport: (1) strain due to lattice mismatch
between Cu and Cu2O and (2) electrostatic interactions. We show that the formation and
diffusion of Cu–O clusters can accelerate the Cu transport process, especially in the
presence of surface vacancy defects. Our atomistic simulations demonstrate that the Cu
atom detachment progresses from the top of the step edge into deeper layers, and the
detachment rate is enhanced with elevated temperatures.
Author
Abhishek Bagusetty, Pabitra Choudhury, Wissam A. Saidi, Bridget Derksen, Elizabeth
Gatto, and J. Karl Johnson
Graphane functionalized with hydroxyl groups is shown to rapidly conduct protons under
anhydrous conditions through a contiguous network of hydrogen bonds. Density functional
theory calculations predict remarkably low barriers to diffusion of protons along a 1D
chain of surface hydroxyls. Diffusion is controlled by the local rotation of hydroxyl
groups, a mechanism that is very different from that found in 1D water wires in confined
nanopores or in bulk water. The proton mean square displacement in the 1D chain was
observed to follow Fickian diffusion rather than the expected single-file mobility. A
charge analysis reveals that the charge on the proton is essentially equally shared by
all hydrogens bound to oxygens, effectively delocalizing the proton.
Nickel-based alloys are widely applied materials in high-temperature applications
because they exhibit superior corrosion resistance and mechanical properties. The
effects of sulfur, which is invariably present in industrial atmospheres, on the early
stages of oxidation of Ni-based surfaces are not well understood. Here we use density
functional theory to investigate the interactions of sulfur, SO, and SO2 with the
Ni(111) and Cr-doped Ni(111) surface and elucidate their electronic interactions and
potential energy surfaces. The results show that Cr doping of the Ni(111) surface
increases the adsorption energies of sulfur, oxygen on the sulfur pre-adsorbed
condition, SO and SO2. Further, this increase positively correlates with Cr
concentration on top of the Ni(111) surface, although sulfur does not have any
preferential interaction with Cr. This explains why Cr doping has little effect on the
activation energy of sulfur for the most preferable diffusion path. Nevertheless, the
increase in adsorption energies indicates a strong interaction with Cr-doped surfaces,
which is due to the Cr-enhanced charge transfer to sulfur adsorbates. The existence of
pre-adsorbed sulfur is shown to have a destabilizing effect on the oxygen interactions
with the surfaces. Our results show that Cr doping helps to stabilize the protective
oxide scale on Ni(111) surfaces and enhances its corrosion resistance.
Author
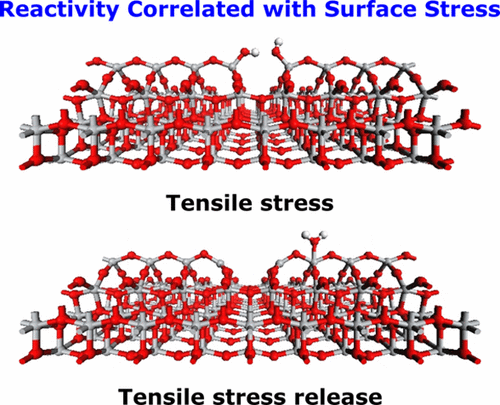
Yongliang Shi, Huijuan Sun, Wissam A. Saidi , Manh Cuong Nguyen, Cai Zhuang Wang,
Kaiming Ho, Jinlong Yang, and Jin Zhao
In contrast with theoretical predictions in which anatase TiO2(001) and its (1 × 4)
reconstructed surfaces are highly reactive, recent experimental results show this
surface to be inert except for the defect sites. In this report, based on a systematic
study of anatase TiO2(001)-(1 × 4) surface using first-principles calculations, the
tensile stress is shown to play a crucial role on the surface reactivity. The predicted
high reactivity based on add-molecule model is due to the large surface tensile stress,
which can be easily suppressed by a stress-release mechanism. We show that various
surface defects can induce stress release concomitantly with surface passivation. Thus
the synthesis of anatase(001) surface with few defects is essential to improve the
reactivity, which can be achieved, for example, via H2O adsorption. Our study provides a
uniform interpretation of controversial experimental observations and theoretical
predictions on anatase TiO2(001) surface and further proposes new insights into the
origin of surface reactivity.
Author
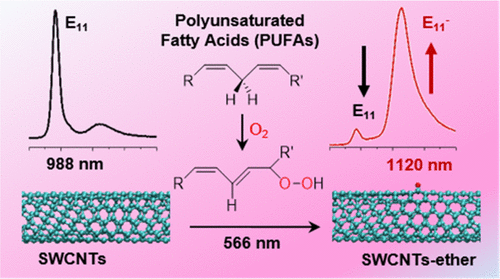
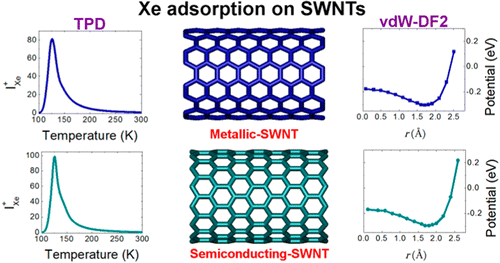
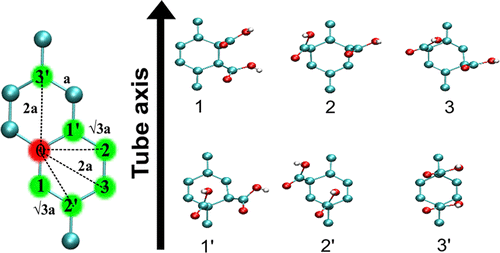
Cheuk Fai Chiu, Wissam A. Saidi, Valerian E. Kagan, and Alexander Star
Single-walled carbon nanotubes (SWCNTs) have been incorporated in many emerging
applications in the biomedical field including chemical sensing, biological imaging,
drug delivery, and photothermal therapy. To overcome inherent hydrophobicity and improve
their biocompatibility, pristine SWCNTs are often coated with surfactants, polymers,
DNA, proteins, or lipids. In this paper, we report the effect of polyunsaturated fatty
acids (PUFAs) on SWCNT photoluminescence. A decrease in the SWCNT bandgap emission (E11)
and a new red-shifted emission (E11-) were observed in the presence of PUFAs. We
attribute the change in SWCNT photoluminescence to the formation of oxygen-containing
defects by lipid hydroperoxides through photo-oxidation. The observed changes in
near-infrared emission of SWCNTs are important for understanding the interaction between
SWCNTs and lipid biocorona. Our results also indicate that photo-excited SWCNTs can
catalyze lipid peroxidation similarly to lipoxygenases.
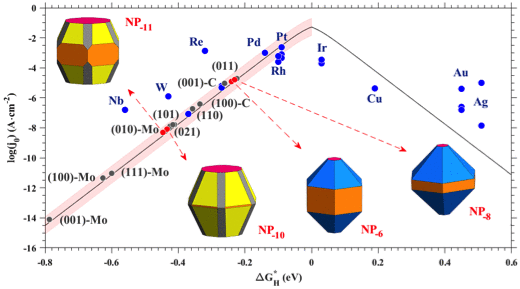
The use of water electrocatalysis for hydrogen production is a promising, sustainable
and greenhouse-gas-free process to develop disruptive renewable energy technologies.
Transition metal carbides, in particular β-phase Mo2C, are garnering increased attention
as hydrogen evolution reaction (HER) catalysts due to their favourable synthesis
conditions, stability and high catalytic efficiency. We use a thermodynamic approach in
conjunction with density functional theory and a kinetic model of exchange current
density to systematically study the HER activity of β-Mo2C under different experimental
conditions. We show that the (011) surface has the highest HER activity, which is
rationalized by its lack of strong Mo-based hydrogen adsorption sites. Thus, the HER
efficiency of β-Mo2C can be tuned using nanoparticles (NPs) that expose larger fractions
of this termination. We give definite maps between NP morphologies and experimental
synthesis conditions, and show that the control of carbon chemical potential during
synthesis can expose up to 90% of (011) surface, while as H2 ambient has little effect
on NPs morphology. The “volcano” plot shows that under these optimum conditions, the NP
exchange current density is ~10-5 A/cm2, that is only slightly smaller than that of Pt
(111).
Author
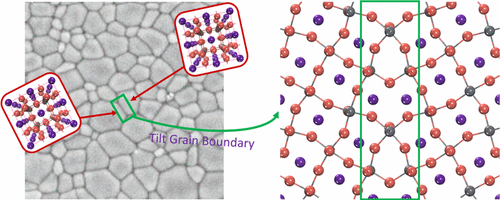
Yaguang Guo, Qian Wang, and Wissam A. Saidi
Organometal trihalide perovskites are emerging as very promising photovoltaic materials,
which is rivaling that of single crystal silicon solar cells despite their
polycrystalline nature with relatively high density of grain boundaries (GBs). There is
a lack of understanding of the effects of GBs on halide perovskites as their presence in
silicon and other photovoltaic materials is generally detrimental to their photovoltaic
properties. Using first-principles calculations, we systematically investigate the
geometric structures of high-angle tilt GBs in halide perovskites CsPbX3 (X = Cl, Br,
and I) starting from the coincidence site lattice model and refining using crystal
shifts and lattice expansion. Electronic density of states calculations reveal that GBs
in halides perovskites do not generate midgap states because of the large distance
between the unsaturated atoms and the atomic reconstructions in the GB region. However,
we show that the GBs can induce different very shallow states near the valence band edge
that can hinder hole diffusion. We further extend the results to MAPbI3 GBs and also
show their benign effect on optoelectronic properties.
Author
Benjamin J. Foley, Justin Girard, Blaire A. Sorenson, Alexander Z. Chen, J. Scott
Niezgoda, Matthew R. Alpert, Angela F. Harper, Detlef-M. Smilgies, Paulette Clancy,
Wissam A. Saidi and Joshua J. Choi
Accelerating the progress toward realizing metal halide perovskite solar cells with
improved efficiency, stability and reliability requires a deeper understanding of the
thin film formation processes. This paper investigates the impact of rationally selected
chemical additives in precursor solutions on the nucleation and growth of metal halide
perovskite thin films. Computational screening was performed to guide the selection of
tetrahydrothiophene oxide as an additive with stronger solvation efficacy than all other
commonly used solvents. In situ grazing incidence X-ray diffraction measurements show
that the additives suppress the formation of homogeneous nuclei as well as crystalline
intermediate structures. Instead, heterogeneous nucleation on the substrate surface and
growth of a thin film with a strongly preferential crystallographic orientation occur
directly from the precursor solution. Density functional theory calculations show that
the crystallographic orientation of the thin films can be tuned by altering the surface
energies with the chemical additives. The crystallographic orientation of the thin films
is found to have a significant impact on the open circuit voltage of solar cell devices,
highlighting the importance of controlling the metal halide perovskite thin film
orientation for improved solar cell efficiency.
Author
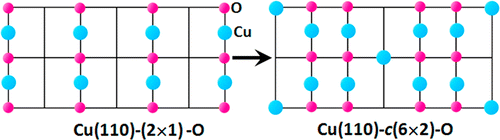
Weitao Shan, Qianqian Liu, Jonathan Li, Na Cai, Wissam A. Saidi, and Guangwen Zhou
Using a combination of scanning tunneling microscopy (STM) and density functional theory
(DFT) modeling, we determine the mechanism of the atomic structural evolution of the
oxygenated Cu(110) surface induced by the reaction of adsorbed hydrogen with chemisorbed
oxygen in the Cu(110)-c(6 × 2)-O structure. Our STM observations show that the
reconstructed Cu(110)-c(6 × 2)-O surface undergoes a phase transition to the (2 × 1)-O
reconstruction in the course of oxygen loss induced by the reaction with H2 gas. Using
DFT modeling, we find that the surface phase transition is initiated via the adsorption
of molecular hydrogen on the chemisorbed oxygen, which results in the formation of H2O
molecules that desorb spontaneously from the surface. The loss of chemisorbed oxygen
induces the c(6 × 2) → (2 × 1) transition that involves the diffusion of Cu―O―Cu chains
along the ⟨1¯10⟩
direction.
Author
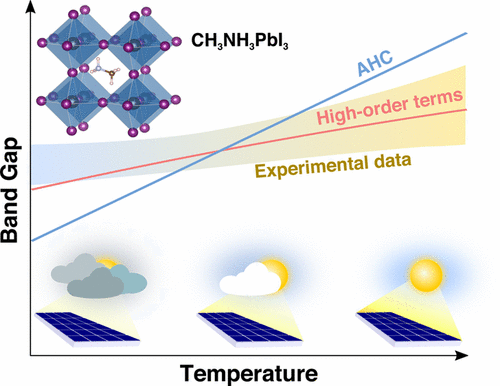
Wissam A. Saidi, Samuel Ponce, and Bartomeu Monserrat
Environmental effects and intrinsic energy-loss processes lead to fluctuations in the
operational temperature of solar cells, which can profoundly influence their power
conversion efficiency. Here we determine from first-principles the effects of
temperature on the band gap and band edges of the hybrid pervoskite CH3NH3PbI3 by
accounting for electron–phonon coupling and thermal expansion. From 290 to 380 K, the
computed band gap change of 40 meV coincides with the experimental change of 30–40 meV.
The calculation of electron–phonon coupling in CH3NH3PbI3 is particularly intricate as
the commonly used Allen–Heine–Cardona theory overestimates the band gap change with
temperature, and excellent agreement with experiment is only obtained when including
high-order terms in the electron–phonon interaction. We also find that spin–orbit
coupling enhances the electron–phonon coupling strength but that the inclusion of
nonlocal correlations using hybrid functionals has little effect. We reach similar
conclusions in the metal–halide perovskite CsPbI3. Our results unambiguously confirm for
the first time the importance of high-order terms in the electron–phonon coupling by
direct comparison with experiment.
Hybrid organic-inorganic perovskites, as well as the perovskites in general, are known
for their phase complexity evidenced by the stabilization of different polymorphs, and
thus an understanding of their regions of stability and transitions can be important for
their photovoltaic and optoelectronic technologies. Here we use a multiscale approach
based on first-principles calculations with van der Waals corrections and classical
force-field molecular dynamics to determine the finite-temperature properties of the
tetragonal and cubic phases of CH3NH3PbI3. Temperature effects are implicitly included
using the quasi-harmonic approximation that can describe anharmonic behavior due to
thermal expansion through the dependence of the harmonic frequencies on structural
parameters. Our finite-temperature free-energy surfaces predict the lattice and elastic
moduli evolution with temperature, and show in particular that the calculated lattice
parameters of the cubic and tetragonal phases are to within 1% of experimental values.
Further, our results show that the phonons are the major contributing factor for
stabilizing the cubic phase at high temperatures mainly due to the low-energy phonon
modes that are associated with the inorganic lattice. On the other hand, the
configurational entropy due to CH3NH3 + rotational degrees of freedom is slightly more
favored in the cubic phase and amounts to less than 0.2% of the T = 0 K free-energy
difference between the two phases.
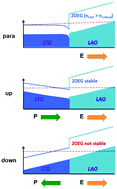
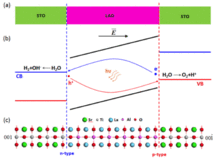
The two-dimensional electron gas (2DEG) formed at the interface between two insulating
materials LaAlO3 (LAO) and SrTiO3 (STO) has recently generated a lot of interest. Here,
based on first-principles density functional theory calculations, we investigate the
existence and stability of the 2DEG under the application of a biaxial strain on the
LAO/STO(001) heterostructure. The compressive strain induces ferroelectric (FE)
polarization in STO, which allows for the tunability of the 2DEG by reversing the STO
polarization orientation. We show that the formation of the 2DEG is unstable when LAO
and STO have the same polarization direction. On the other hand, the 2DEG will always
form if the two polarizations are in the opposite directions regardless of the LAO
thickness, which is in contrast to the unstrained interface that has a critical
thickness for stabilizing the 2DEG. We show that the underpinnings of this behavior are
due to charge passivation and band gap alignment.
Author Weibin Chu, Wissam A. Saidi, Qijing Zheng, Yu Xie,
Zhenggang Lan, Oleg V. Prezhdo, Hrvoje Petek, and Jin Zhao
Photogenerated charge carrier dynamics near molecule/TiO2 interfaces are important for
the photocatalytic and photovoltaic processes. To understand this fundamental aspect, we
performed a time-domain ab initio nonadiabatic molecular dynamics study of the
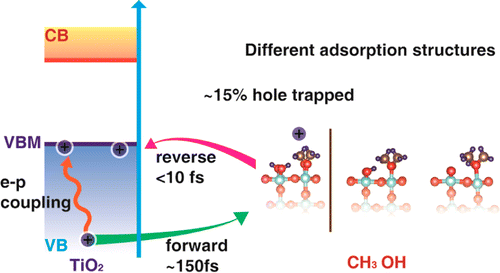
photogenerated hole dynamics at the CH3OH/rutile TiO2(110) interface. We studied the
forward and reverse hole transfer between TiO2 and CH3OH as well as the hole energy
relaxation to the valence band maximum. First, we show that the hole-trapping ability of
CH3OH depends strongly on the adsorption structure. Only when the CH3OH is deprotonated
to form chemisorbed CH3O will ∼15% of the hole be trapped by the molecule. Second, we
find that strong fluctuations of the HOMO energies of the adsorbed molecules induced by
electron-phonon coupling provide additional channels, which accelerate the hole energy
relaxation. Third, we demonstrate that the charge transfer and energy relaxation
processes depend significantly on temperature. When the temperature decreases from 100
to 30 K, the forward hole transfer and energy relaxation processes are strongly
suppressed because of the reduction of phonon occupation. These results indicate that
the molecule/TiO2 energy level alignment, thermal excitation of a phonon, and
electron-phonon coupling are the key factors that determine the photogenerated hole
dynamics. Our studies provide valuable insights into the photogenerated charge and
energy transfer dynamics at molecule/semiconductor interfaces.
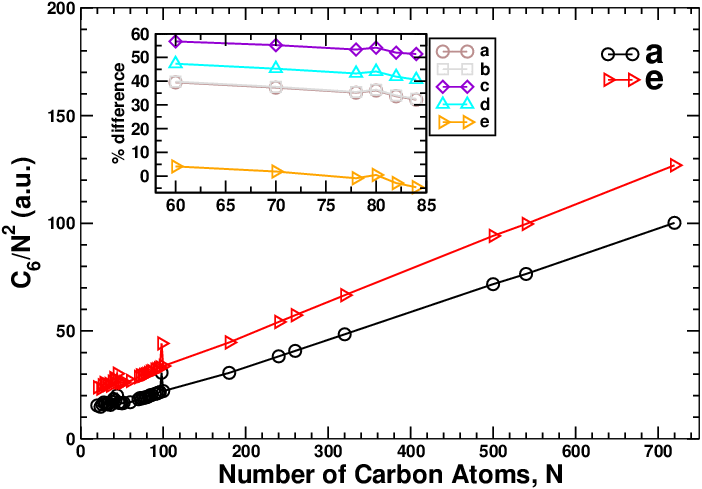
The van der Waals C6 coefficients of fullerenes are shown to exhibit an anomalous
dependence on the number of carbon atoms N such that C 6 ∝ N 2.2 as predicted using
state-of-the-art quantum mechanical calculations based on fullerenes with small sizes,
and N 2.75 as predicted using a classical-metallic spherical-shell approximation of the
fullerenes. We use an atomistic electrodynamics model where each carbon atom is
described by a polarizable object to extend the quantum mechanical calculations to
larger fullerenes. The parameters of this model are optimized to describe accurately the
static and complex polarizabilities of the fullerenes by fitting against accurate ab
initio calculations. This model shows that C 6 ∝ N 2.8, which is supportive of the
classical-metallic spherical-shell approximation. Additionally, we show that the
anomalous dependence of the polarizability on N is attributed to the electric charge
term, while the dipole–dipole term scales almost linearly with the number of carbon
atoms.
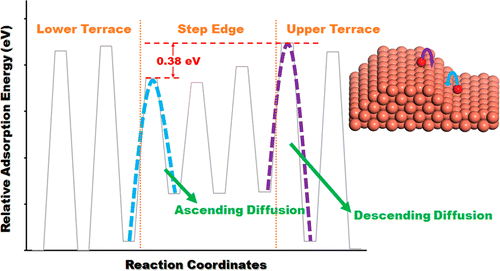
Metal surface oxidation is governed by surface mass transport processes. Realistic
surfaces have many defects such as step edges, which often dictate the oxide growth
dynamics and result in novel oxide nanostructures. Here we present a comprehensive and
systematic study of the oxidation of stepped (100), (110) and (111) Cu surfaces using a
multiscale approach employing density functional theory (DFT) and reactive force field
molecular dynamics (MD) simulations. We show that the early stages of oxidation of these
stepped surfaces can be qualitatively understood from the potential energy surface of
single oxygen adatoms, namely, adsorption energies and Ehrlich-Schwöbel barriers. These
DFT predictions are then validated using classical MD simulations with a newly optimized
ReaxFF force field. In turn, we show that the DFT results can be explained using a
simple bond-counting argument that makes our results general and transferable to other
metal surfaces.
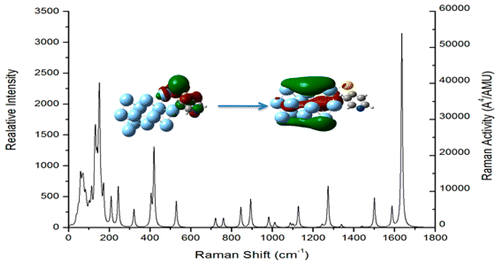
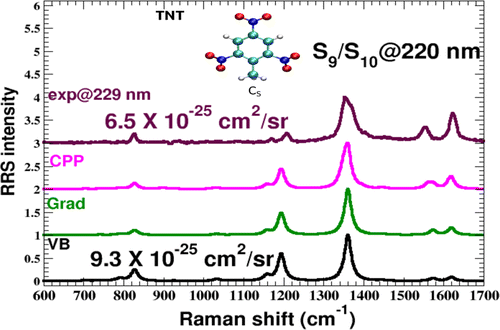
Author Ronald L. Birke, John R. Lombardi, Wissam A. Saidi, and
Patrick Norman
We have used time-dependent density functional theory in conjunction with the CAM-B3LYP
functional and MWB28/aug-cc-pVDZ basis set to determine non-, near-, and on-resonance
Raman spectra for a complex formed by 4-mercaptopyridine (4-Mpy) binding with a Ag13
cluster via the thiolate Ag–S bond. Geometry optimizations of the Ag13-4-Mpy complex
showed an on-top structure directly bound to one Ag atom with the ring of the molecule
almost flat with respect to two Ag atoms of the complex. The corresponding
B3LYP/MWB28/aug-cc-pVDZ geometry is also an on-top structure directly bound to one Ag
atom, but the molecule is directed away from the surface. The near-resonance Raman
calculations were carried out in the infinite lifetime approximation, while the
on-resonant Raman excitation profiles were calculated with the complex polarization
propagator (CPP) approach, introducing a half width at half-maximum spectral broadening
of 0.2 eV. Calculation of the UV–vis spectra of the isolated 4-Mpy and of the Ag13-4-Mpy
complex showed that binding shifts the spectra from deep in the UV to the visible
region. Calculation of the near-resonance Raman spectra of the two structures of the
complex at 410 (3.025 eV) and 425 nm (2.918 eV) showed a strong enhancement. A very
large variation across vibrational modes by a factor of at least 103 was found for both
the static chemical enhancement and charge-transfer (CT) enhancement mechanisms. This
large variation in enhancement factor indicates that B-term Herzberg–Teller scattering
is occurring because inactive or very low intensity modes in the static spectra of the
molecule are much stronger in both the static and near-resonance spectra of the complex.
From the excitation profile using the CPP method, an overall surface enhancement on the
order 103 or higher was found for individual modes on excitation into a CT excited
state.
Author Qing Zhu, Lianfeng Zou, Guangwen Zhou, Wissam A. Saidi, and
Judith C. Yang
Understanding of metal oxidation is critical to corrosion control, catalysis synthesis,
and advanced materials engineering. Although, metal oxidation process is rather
complicated, different processes, many of them coupled, are involved from the onset of
reaction. Since first introduced, there has been great success in applying
heteroepitaxial theory to the oxide growth on a metal surface as demonstrated in the Cu
oxidation experiments. In this paper, we review the recent progress in experimental
findings on Cu oxidation as well as the advances in the theoretical simulations of the
Cu oxidation process. We focus on the effects of defects such as step edges, present on
realistic metal surfaces, on the oxide growth dynamics. We show that the surface steps
can change the mass transport of both Cu and O atoms during the oxide growth, and
ultimately lead to the formation of different oxide morphology. We also review the
oxidation of Cu alloys and explore the effect of secondary element to the oxide growth
on a Cu surface. From the review of the work on Cu oxidation, we demonstrate the
correlation of theoretical simulations at multiple scales with various experimental
techniques.
Author Hongli Guo, Wissam A Saidi, Jinlong Yang and Jin Zhao
We propose that a nano-scale thin film based on polar-nonpolar transition-metal oxide
heterostructure can be used as a highly-efficient photocatalyst. This is demonstrated
using a SrTiO3/LaAlO3/SrTiO3 sandwich-like heterostructure with a photocatalytic
activity in the near-infrared region using first principles calculations. The effect of
the polar nature of LaAlO3 is two-fold. First, the induced electrostatic field
accelerates the photo-generated electrons and holes into opposite directions and
minimizes their recombination rates. Hence, the reduction and oxidation reactions can be
instigated at the SrTiO3 surfaces located on opposite sides of the heterostructure.
Second, the electric field reduces the band gap of the system making it photoactive in
the infrared region. We also show that charge separation can be enhanced in using
compressive strain engineering that creates a ferroelectric instability in STO. The
proposed setup is ideal for tandem oxide photocatalysts especially when combined with
photoactive polar materials.
Author Benjamin J Foley, Daniel L Marlowe, Keye Sun, Wissam A
Saidi, Louis Scudiero, Mool C Gupta, and Joshua J Choi
Temperature dependent energy levels of methylammonium lead iodide are investigated using
a combination of ultraviolet photoemission spectroscopy and optical spectroscopy. Our
results show that the valence band maximum and conduction band minimum shift down in
energy by 110 meV and 77 meV as temperature increases from 28 °C to 85 °C. Density
functional theory calculations using slab structures show that the decreased orbital
splitting due to thermal expansion is a major contribution to the experimentally
observed shift in energy levels. Our results have implications for solar cell
performance under operating conditions with continued sunlight exposure and increased
temperature.
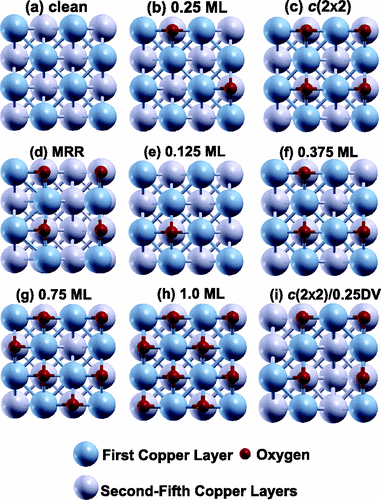
Contacts between metal surfaces and MoS2 are crucial for the utilization of
MoS2 in different technologies. Here we systematically investigate using
first-principles density functional theory the adsorption and diffusion on
MoS2(001) of a wide range of metals from Groups I–IV in addition to all of
the 3d transition metals (TMs) and selected 4d and 5d TMs. The binding mechanisms as
well as trends in the binding energies are elucidated by examining the electronic
structure of the system, and in particular the interplay between Coulomb interactions,
Pauli repulsion, and ndm(n +
1)sx → ndm+1(n +
1)sx–1 (x = 1, 2; n = 3, 4, 5) promotion
energies. We show that the metal-induced workfunction reduction is correlated with the
ionization potential of the isolated atom and is furthermore linearly dependent on the
interfacial dipole moment with an offset term. Additionally, the growth morphologies of
the metal nanoparticles on MoS2 are predicted by analyzing the monomer
adhesion energy and its mobility on the substrate. Our results are in line with recent
experiments showing that Ag and Au follow a Volmer–Weber growth mode on
MoS2(001).
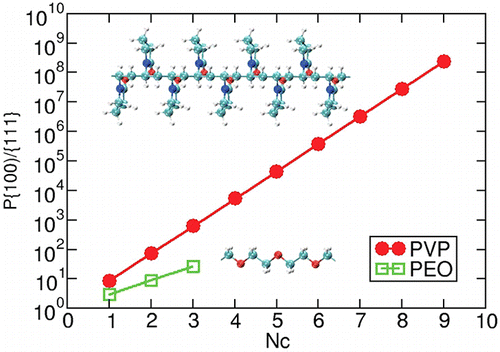
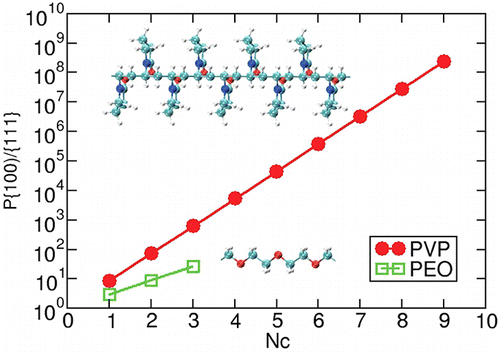
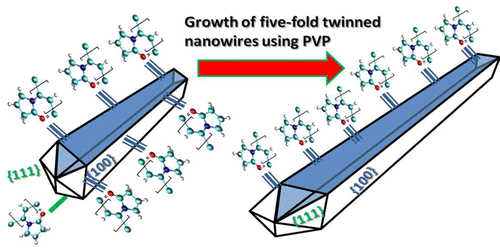
Authors Shih-Hsien Liu, Wissam A. Saidi, Ya Zhou, and Kristen A.
Fichthorn
We use density-functional theory (DFT) and molecular dynamics (MD) to resolve the role
of polyvinylpyrrolidone (PVP) in the shape-selective synthesis of Au nanostructures.
Using DFT, we probe the adsorption-induced surface energies and spatially resolved
binding of PVP monomer analogs on Au(111), Au(100), and (5 × 1) Au(100)-hex. These
calculations suggest that {111} facets should be prevalent in Au nanostructures grown
with the help of PVP. We explore the role of solvent and find that, while solvent
weakens binding, it does not change the trends we observe in vacuum. We fit an ad hoc
interatomic potential to the DFT results so we can describe the binding of PVP to the Au
surfaces. Using MD simulations based on this potential, we investigate the PVP-induced
surface energies, PVP binding affinities, and oxygen density profile of atactic PVP
icosamers on Au(111) and (5 × 1) Au(100)-hex. We conclude that {111}-faceted Au
nanocrystals are preferred in PVP-mediated synthesis of Au nanostructures. The
reconstruction of Au(100) is important in achieving {111}-facet selectivity.
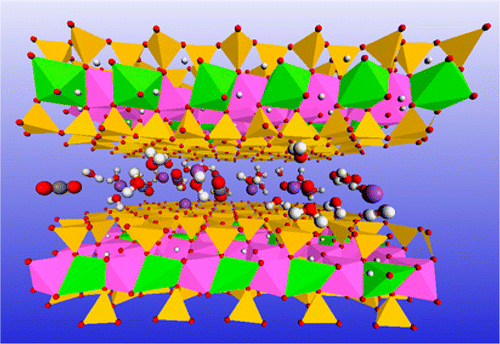
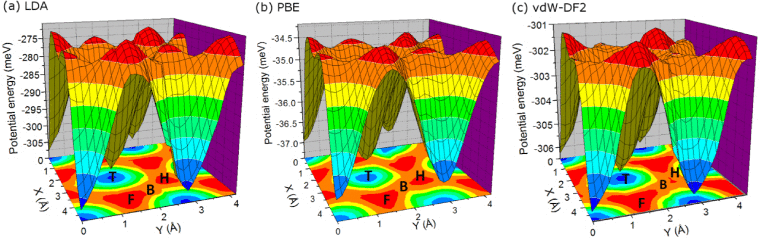
Authors Y. Qi, J. M. P. Martirez, Wissam A. Saidi, J. J. Urban, W.
S. Yun, J. E.Spanier, and A. M. Rappe
We investigate the origin of the depolarization rates in ultrathin adsorbate-stabilized
ferroelectric wires. By applying density functional theory calculations and analytic
modeling, we demonstrate that the depolarization results from the leakage of charges
stored at the surface adsorbates, which play an important role in the polarization
stabilization. The depolarization speed varies with thickness and temperature, following
several complex trends. A comprehensive physical model is presented, in which quantum
tunneling, Schottky emission, and temperature-dependent electron mobility are taken into
consideration. This model simulates experimental results, validating the physical
mechanism. We also expect that this improved tunneling-Schottky emission model could be
applied to predict the retention time of polarization and the leakage current for
various ferroelectric materials with different thicknesses and temperatures.
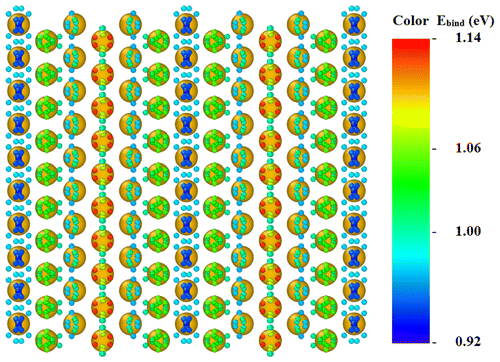
The dispersion of Pt metallic nanoparticles on different supports is of high relevance
for designing more efficient and less expensive catalysts. In order to understand the
nucleation and epitaxial growth of Pt nanoparticles and thin films on MoS2
monolayers, we have systematically analyzed, by first-principles density functional
calculations, the evolution of morphology and atomic structure of supported
(Pt)n nanoparticles (NPs) on MoS2(001) for n ≤ 12.
We find that n = 5 is the cluster size where the growth of the NPs transforms
from two- to three-dimensional (2D to 3D). Owing to the topography of
MoS2(001), the 2D NPs mostly attach to the support via direct bonding with Mo
atoms that sit in the troughs of the surface, while the 3D NPs are bonded to the sulfur
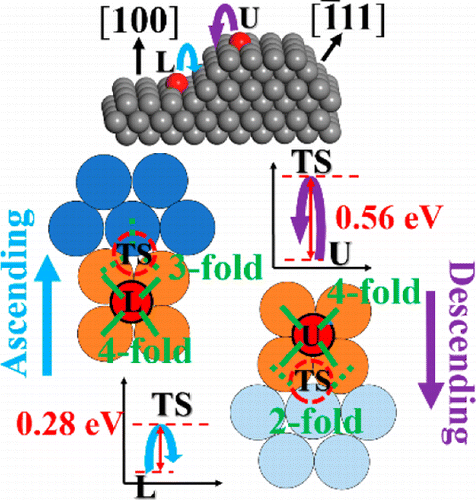
atoms that are more extended in the vacuum region. Furthermore, we find that Pt is
sufficiently mobile on the surface where the number of hopping events per second is
≈103 s–1 along [101̅] and ≈10 s–1 along [11̅0] at room
temperature. The somewhat large mobility suggests that monomer diffusion is not likely
to be the rate-limiting step for Oswald ripening and that Pt sputtering on
MoS2(001) will result in relatively large particles rather than a fine
dispersion. The existence of a fast diffusion channel along [101̅] suggests that the
morphology of the NPs is anisotropic.
Authors Qing Zhu, Wissam A. Saidi, and Judith C. Yang
Surface defects such as step edges play an important role in determining the surface
properties, affecting immensely the growth mechanisms and morphologies of the
nanostructures in epitaxial film growth processes. Here, we probe the dynamics of the
oxidation on stepped Cu(100) using molecular dynamic simulations in conjunction with a
reactive force field, and we elucidate the mechanisms and energy barriers affecting the
oxidation process. Molecular dynamic simulations show that the adsorbed oxygen adatoms
are unevenly distributed on the stepped surface, favoring the top terrace. We show that
this behavior is due to Ehrlich–Schwöbel (ES) barrier effect. However,
differently from the reduced interlayer self-diffusion in descending a step as in a
conventional ES barrier effect, we find instead that the ES barrier reduces the
ascending diffusion barrier for oxygen, promoting its transport across the step edge and
enhancing oxidation of the upper terrace. Additionally, we find that the ES barrier is
step-height dependent, where higher step edges reduce more the oxygen-ascending
diffusion barrier and favor more oxidation of the upper terraces of stepped
surfaces.
Authors Kevin D Parrish, Ankit Jain, Jason M Larkin, Wissam A
Saidi, Alan JH McGaughey
The strain-dependent phonon properties and thermal conductivities of a soft system
[Lennard-Jones (LJ) argon] and a stiff system (silicon modeled using first-principles
calculations) are predicted using lattice dynamics calculations and the Boltzmann
transport equation. As is commonly assumed for materials under isotropic strain, the
thermal conductivity of LJ argon decreases monotonically as the system moves from
compression into tension. The reduction in thermal conductivity is attributed to
decreases in both the phonon lifetimes and group velocities. The thermal conductivity of
silicon, however, is constant in compression and only begins to decrease once the system
is put in tension. The silicon lifetimes show an anomalous behavior, whereby they
increase as the system moves from compression into tension, which is explained by
examining the potential energy surface felt by an atom. The results emphasize the need
to separately consider the harmonic and anharmonic effects of strain on material
stiffness, phonon properties, and thermal conductivity.
Authors Wissam A. Saidi, John Mark P. Martirez, and Andrew M.
Rappe
We present a systematic evaluation of the effects of polarization switchability on
surface structure and stoichiometry in BaTiO3 and PbTiO3
ferroelectric oxides. We show that charge passivation, mostly by ionic surface
reconstructions, is the driving force for the stability of the surfaces, which suggests
that varying the substrate polarization offers a new mechanism for controlling surface
reconstructions in polar systems and inducing highly nonstoichiometric structures.
Conversely, for thin-films the chemical environment can drive polarization switching via
induced compositional changes on the surface. We find that the value of the oxygen
partial pressure for the positive-to-negative polar transition is in good agreement with
the recent experimental value for thin-film PbTiO3. For BaTiO3, we
show that it is harder for oxygen control to drive polar transition because it is more
difficult to reduce. This study opens up the possibility of real-time control of
structure and composition of oxide surfaces.
Authors Liang Li, Na Cai, Wissam A Saidi, Guangwen Zhou
Surface steps are typically assumed as a source of adatoms for oxygen-chemisorption
induced surface reconstruction, but few microscopic observations have been made in the
vicinity of steps on reconstructing surfaces. Using in situ scanning tunneling
microscopy, we provide direct evidence that surface steps are the source of Cu adatoms
for the Cu(1 1 0)single bond(2 × 1)-O restructuring. Using density functional
theory, we show that the role of oxygen is to stabilize Cu adatoms detached from step
edges via the barrier-less formation of Cusingle bondO dimers on terraces. Incorporating
this atomic process of capturing Cu adatoms into kinetic Monte Carlo simulations
reproduces the experimentally observed (2 × 1)-O reconstruction.
Authors Qianqian Liua, Liang Lia, Na Caia, Wissam A. Saidib,
Guangwen Zhou
From an interplay between variable temperature scanning tunneling microscopy and
density–functional theory calculations, the evolution of oxygen chemisorption-induced
surface reconstructions of the Cu(110) surface is determined. The surface
reconstructions proceed via a sequential pathway with increasing oxygen surface
coverage. The (2 × 1) reconstruction occurs first and then transits to the c(6
× 2) phase with a higher oxygen coverage through a mechanism that consumes the
existing (2 × 1) phase with the supply of Cu adatoms from step edges and terraces.
The temperature dependence of the (2 × 1) ? c(6 × 2) transition demonstrates
that the surface phase transition is an activated process for breaking up added
Cu–O–Cu rows in the (2 × 1) structure. Comparison between the experimental
observations and the theoretical surface phase diagram obtained from first-principles
thermodynamic calculations reveals that the (2 × 1) ? c(6 × 2) transition
takes place at the oxygen chemical potentials that are far above the chemical potential
for Cu2O bulk oxide formation, reflecting the existence of kinetic limitations to the
surface phase transition and the bulk oxide formation.
Authors Liang Li, Langli Luo, Jim Ciston, Wissam A. Saidi, Eric A.
Stach, Judith C. Yang, and Guangwen Zhou
We report in situ atomic-resolution transmission electron microscopy observations of
the oxidation of stepped Cu surfaces. We find that the presence of surface steps both
inhibits oxide film growth and leads to the oxide decomposition, thereby resulting in
oscillatory oxide film growth. Using atomistic simulations, we show that the oscillatory
oxide film growth is induced by oxygen adsorption on the lower terrace along the step
edge, which destabilizes the oxide film formed on the upper terrace.
MoS2 and other transition metal dichalcogenides are considered as
potential materials in many applications including future electronics. A prerequisite
for these applications is to understand the nature of the MoS2 contact with
different metals. We use semi-local density functional theory in conjunction with
dispersion corrections to study the heterostructures composed of Pd and Pt monolayers
with (111) orientation grown pseudomorphically on MoS2(001). The interface
properties are mapped as a function of the number of deposited overlayers, as well as a
function of tensile and compressive strains. Although we show that the dependence of the
contacts on strain can be fully explained using the d-band model, we find that their
evolution with the number of deposited metal layers is markedly different between Pd and
Pt, and at variance with the d-band model. Specifically, the Pt/MoS2
heterostructures show an anomalous large stability with the deposition of two metal
monolayers for all investigated strains, while Pd/MoS2 exhibits a similar
behavior only for compressive strains. It is shown that the results can be rationalized
by accounting for second-nearest-neighbor effect that couples MoS2 with the
subsurface metal layers. The underpinnings of this behavior are attributed to the larger
polarizability and cohesive energy of Pt compared to Pd, that leads to a larger
charge-response in the subsurface layers.
The stability and the electronic structure of layered heterostructures MX2 (M = Mo
or W and X = S or Se) and graphene (GA) are systematically investigated using
first-principles methods. The calculations cover pristine and defected GA systems with
up to 12% nitrogen substitutional defects. It is found that the van der Waals (vdW)
epitaxy of MX2 on undoped GA substrate, whether pristine or defected, follows a
Volmer–Weber growth-mode resulting in thick MX2 films. On the other hand, nitrogen
doping of pristine GA (N-GA) and also of GA with Stone–Wales (SW) defects increases
the MX2/GA heterostructure adhesion energy favoring the growth of ultrathin MX2 layers.
This growth-mode change in MoS2 due to nitrogen doping is in agreement with
recent experiments. Furthermore, our study demonstrates that the yield of ultrathin MX2
films can be increased if the N-GA samples have a larger concentration of SW defects or
nitrogen. The underpinnings of the extra stability of these N-GA substrates are due to
charge-transfer effects that decrease the Pauli repulsion between the two layered
systems.
Authors Qing Zhu, Chris Fleck, Wissam A. Saidi, Alan McGaughey,
Judith C. Yang
A three dimensional (3D) kinetic Monte Carlo (KMC) code has been developed that
simulates the general behavior of the 3D irreversible nucleation and growth of epitaxial
islands, as motivated by experimental observations of oxide nuclei formation and growth
during the early stages of copper oxidation. This package was originally a versatile two
dimensional (2D) KMC code [Thin Film Oxidation (TFOx)] that considered a variety of
elementary steps, including deposition, adsorption, surface diffusion, aggregation,
desorption, and substrate-mediated indirect interactions between static adatoms. We
extended TFOx to describe 3D island growth. This new version of TFOx is composed of a
C++ console program and Python graphical user interface (GUI), such that parameterized
simulation, parallel execution, and 3D growth capabilities are feasible. We examined the
effects of the potential gradient and the Ehrlich–Schwöbel barrier and found that
the 3D island morphology is significantly influenced by the incorporation of these two
factors.
Authors Liang Li, Qianqian Liu, Jonathan Li, Wissam A. Saidi, and
Guangwen Zhou
Oxygen chemisorption induced surface reconstructions are widely observed, but the
atomic processes leading to transitions among oxygen chemisorbed phases are largely
unknown. Using ab initio molecular dynamics and density-functional theory, we study the
kinetic process of the Cu(110)-(2 × 1) ? c(6 × 2) phase transition upon
increasing oxygen surface coverage. We show that the phase transition involves initially
Cu–O dimer and Cu–O–Cu trimer formation with a kinetic barrier of ?0.13 eV,
followed by a barrierless process of forming a four Cu–O–Cu–O chains configuration
that transitions to the c(6 × 2) reconstruction via concerted movement of three Cu
atoms with an associated energy barrier of ?1.41 eV. The larger kinetic barrier is
suggested as the origin of the kinetic hindrance that is inferred from the significant
discrepancy between the experimentally observed temperature and pressure dependent (2
× 1) ? c(6 × 2) phase transition and the equilibrium thermodynamics
prediction.
Authors Hao Bai, Wentao Jiang, Gregg P. Kotchey, Wissam A. Saidi,
Benjamin J. Bythell, Jacqueline M. Jarvis, Alan G. Marshall, Renã A. S.
Robinson, and Alexander Star
Graphene represents an attractive two-dimensional carbon-based nanomaterial that
holds great promise for applications such as electronics, batteries, sensors, and
composite materials. Recent work has demonstrated that carbon-based nanomaterials are
degradable/biodegradable, but little work has been expended to identify products formed
during the degradation process. As these products may have toxicological implications
that could leach into the environment or the human body, insight into the mechanism and
structural elucidation remain important as carbon-based nanomaterials become
commercialized. We provide insight into a potential mechanism of graphene oxide
degradation via the photo-Fenton reaction. We have determined that after 1 day of
treatment intermediate oxidation products (with MW 150–1000 Da) were generated. Upon
longer reaction times (i.e., days 2 and 3), these products were no longer present in
high abundance, and the system was dominated by graphene quantum dots (GQDs). On the
basis of FTIR, MS, and NMR data, potential structures for these oxidation products,
which consist of oxidized polycyclic aromatic hydrocarbons, are proposed.
Authors Erie H. Morales, John Mark P. Martirez, Wissam A. Saidi,
Andrew M. Rappe, and Dawn A. Bonnell
Coexistence of surface reconstructions is important due to the diversity in kinetic
and thermodynamic processes involved. We identify the coexistence of kinetically
accessible phases that are chemically identical and form coherent interfaces. Here, we
establish the coexistence of two phases, c(2 × 2) and c(4 × 4), in
BaTiO3(001) with atomically resolved Scanning Tunneling Microscopy (STM).
First-principles thermodynamic calculations determine that TiO adunits and clusters
compose the surfaces. We show that TiO diffusion results in a kinetically accessible c(2
× 2) phase, while TiO clustering results in a kinetically and thermodynamically
stable c(4 × 4) phase. We explain the formation of domains based on the diffusion
of TiO units. The diffusion direction determines the observed 1D coherent interfaces
between c(2 × 2) and c(4 × 4) reconstructions. We propose atomic models for
the c(2 × 2), c(4 × 4), and 1D interfaces.
Authors Yu Li Huang, Elisabeth Wruss, David A. Egger, Satoshi
Kera, Nobuo Ueno, Wissam A. Saidi, Tomas Bucko, Andrew T.S. Wee and Egbert Zojer
Phthalocyanines are an important class of organic semiconductors and, thus, their
interfaces with metals are both of fundamental and practical relevance. In the present
contribution we provide a combined theoretical and experimental study, in which we show
that state-of-the-art quantum-mechanical simulations are nowadays capable of treating
most properties of such interfaces in a quantitatively reliable manner. This is shown
for Cu-phthalocyanine (CuPc) and Zn-phthalocyanine (ZnPc) on Au(111) and Ag(111)
surfaces. Using a recently developed approach for efficiently treating van der Waals
(vdW) interactions at metal/organic interfaces, we calculate adsorption geometries in
excellent agreement with experiments. With these geometries available, we are then able
to accurately describe the interfacial electronic structure arising from molecular
adsorption. We find that bonding is dominated by vdW forces for all studied interfaces.
Concomitantly, charge rearrangements on Au(111) are exclusively due to Pauli pushback.
On Ag(111), we additionally observe charge transfer from the metal to one of the
spin-channels associated with the lowest unoccupied ?-states of the molecules. Comparing
the interfacial density of states with our ultraviolet photoelectron spectroscopy (UPS)
experiments, we find that the use of a hybrid functionals is necessary to obtain the
correct order of the electronic states. - See more at:
http://www.mdpi.com/1420-3049/19/3/2969/htm#sthash.dOxH6z7Z.dpuf
Authors Ya Zhou, Wissam A. Saidi, and Kristen A. Fichthorn
Polyvinylpyrrolidone (PVP), ethylene glycol (EG), and polyethylene oxide (PEO) are
key molecules in the solution-phase synthesis of Ag nanostructures. To resolve various
aspects of this synthesis, we develop a classical force field to describe the
interactions of these molecules with Ag surfaces. We parametrize the force field through
force and energy matching to results from first-principles density-functional theory
(DFT). Our force field reproduces the DFT binding energies and configurations of these
molecules on Ag(100) and Ag(111). Our force field also yields a binding energy for EG on
Ag(110) that is in agreement with experiment. Molecular-dynamics simulations based on
this force field indicate that the preferential binding affinity of the chains for
Ag(100) increases significantly beyond the segment binding energy for PVP decamers, but
not for PEO. This agrees with experimental observations that PVP is a more successful
structure-directing agent than is PEO.
Authors Ozan Karalti, Xiaoge Su, Wissam A. Al-Saidi, Kenneth D.
Jordan
We present a two-channel dispersion-corrected atom-centered potential (DCACP) method
for correcting BLYP and PBE density functionals for long-range dispersion. The approach,
designated DCACP2, is tested on the S22X5 test set and on isomers of the water hexamer.
The DCACP2 method provides a significantly improved description of the interaction
energies at distances beyond Req than does the single-channel DCACP procedure.
The optical properties, including UV-vis spectra and resonance Raman profiles, of
pristine and defected single-walled carbon nanotubes (SWCNTs) are computed using
state-of-the-art time-dependent density functional theory (TDDFT) as implemented using
the Liouville–Lanczos approach to linear-response TDDFT. The CNT defects were of the
form of Stone–Wales and diatom-vacancies. Our results are in very good agreement with
experimental results where defects were introduced into a part of defect-free CNTs. In
particular, we show that the first and second ?–?* excitation energies are barely
shifted due to the defects and associated with a relatively small reduction in the
maxima of the absorption bands. In contrast, the resonance Raman spectra show close to
an order of magnitude reduction in intensities, offering a means to distinguish between
pristine and defected SWCNTs even at low defect concentrations.
Defects are ubiquitous in carbon nanotubes (CNTs), despite their large formation
energies, and have astounding effects on their physicochemical properties. In this
study, we employ density-functional theory (DFT) calculations to study systematically
the atomic structure, stability, and characteristic vibrations of pristine and defected
zigzag CNTs, where the defects are of the form of Stone–Wales (SW) and diatom
vacancies (DV). The DFT optimized structures and the phonon modes are subsequently used
in conjunction with a semiempirical bond-polarization model to study the nonresonant
Raman spectra. For each defect type, we find two CNT structures with defects parallel or
oblique to the tube axis. For the SW defects, the two structures have similar formation
energies, whereas for the DV defect, only defects parallel to the tube axis are likely
to exist. The results show that the defects induce a blue shift in the radial breathing
mode (RBM) of metallic CNTs, whereas this mode is not shifted for semiconducting CNTs.
However, the RBM shift or its Raman profile is not sensitive to the defect type. The
G-band showed more sensitivity to the defects in the form of a red/blue shift in the
frequency, or a partial/complete defragmentation of the G bands.
First-principles investigations of the electrocatalytic activity toward the
four-electron oxygen reduction-reaction in N-doped graphene quantum dots reveal that
pyridinic and graphitic nitrogen are the most active sites with overpotentials of 0.55
and 0.79–0.90 V, respectively. This agrees with experimental findings. Our
calculations account for van der Waals interactions, solvent effects, and describe the
electrochemistry using standard hydrogen electrode model. The results show correlations
between OH*, OOH*, and O* binding energies that impose a lower limit on the oxygen
reduction overpotential.
Authors Yifan Tang, Seth C. Burkert, Yong Zhao, Wissam A. Saidi,
and Alexander Star
Nitrogen-doped and undoped carbon nanotubes (CNTs) were synthesized from ferrocene,
nickelocene, and cobaltocene metal catalysts. Electrochemical testing for an oxygen
reduction reaction (ORR) showed that nitrogen-doped CNTs synthesized from ferrocene had
improved catalytic activity while nanotubes synthesized from nickelocene and
cobaltocene, doped with a comparable amount of nitrogen and having similar stacked-cups
structure as nitrogen doped CNTs from ferrocene, had a performance only slightly better
than that of undoped CNTs. Ferrocene-based nitrogen-doped CNTs also demonstrated similar
long-term stability and higher CO tolerance compared to Pt/C catalyst. Detailed ORR
mechanisms were also studied and carbon nanomaterials showed different ORR processes as
a result of the metal catalyst utilized in the chemical synthesis. Nitrogen-doped and
undoped CNTs synthesized from nickelocene show a preferential 4-electron process as
compared to materials synthesized from ferrocene and cobaltocene. We believe that the
metal used in the growth process regulates the mechanism of oxygen reduction and can be
used to develop improved nitrogen-doped carbon nanomaterials as nonprecious-metal
catalysts for fuel cells.
Authors Ping Li, Lin Yu, Michael A. Matthews, Wissam A. Saidi, and
J. Karl Johnson
We report a theoretical investigation of H2O adsorption on the NaBH4(100) surface
based on first principles density functional theory with inclusion of dispersion
corrections in order to explore the initial stages of deliquescence at the molecular
level. In the zero coverage limit, H2O is found to bind strongly to sodium sites on
NaBH4(100) through O··· Na and O–H···H–B attractions.
As the coverage increases H2O molecules adsorb on boron sites. H atoms in the adsorbed
H2O monomer adopt tilted down (15°â€“20°) configurations with respect to the
NaBH4(100) surface, which undergoes reconstruction in response to adsorbed H2O by
rotations of BH4– groups of up to 90° and slight distortions of the positions of
Na+ and BH4–. The adsorption energy per H2O is roughly independent of water coverage
up to at least a coverage of four monolayers, suggesting that it is energetically
feasible for water to condense on the surface, in agreement with experiments. We have
experimentally studied the deliquescence of a mixture of NaBH4 with 10 wt % CoCl2. We
found that CoCl2 lowers the deliquescence temperature compared to that for pure NaBH4 at
a given vapor phase mole fraction of water; i.e., the deliquescence relative humidity is
increased because of addition of CoCl2. Thus, while CoCl2 is a catalyst for aqueous
phase hydrolysis of NaBH4, it actually inhibits deliquescence and hence delays the onset
of steam hydrolysis.
Authors Cheuk Fai Chiu, Brian A. Barth, Gregg P. Kotchey, Yong
Zhao, Kristy A. Gogick, Wissam A. Saidi, Stéphane Petoud, and Alexander Star
In this work, we studied enzyme-catalyzed oxidation of single-walled carbon
nanotubes (SWCNTs) produced by the high-pressure carbon monoxide (HiPco) method. While
oxidation via strong acids introduced defect sites on SWCNTs and suppressed their
near-infrared (NIR) fluorescence, our results indicated that the fluorescence of SWCNTs
was restored upon enzymatic oxidation, providing new evidence that the reaction
catalyzed by horseradish peroxidase (HRP) in the presence of H2O2 is mainly a
defect-consuming step. These results were further supported by both UV–vis–NIR and
Raman spectroscopy. Therefore, when acid oxidation followed by HRP-catalyzed enzyme
oxidation was employed, shortened (<300 nm in length) and NIR-fluorescent SWCNTs were
produced. In contrast, upon treatment with myeloperoxidase, H2O2, and NaCl, the oxidized
HiPco SWCNTs underwent complete oxidation (i.e., degradation). The shortened,
NIR-fluorescent SWCNTs resulting from HRP-catalyzed oxidation of acid-cut HiPco SWCNTs
may find applications in cellular NIR imaging and drug delivery systems.
Authors Evgeniy M. Myshakin, Wissam A. Saidi , Vyacheslav N.
Romanov , Randall T. Cygan, and Kenneth D. Jordan
Molecular dynamics simulations using classical force fields were carried out to
study the structural and transport properties of clay mineral–water–CO2 systems at
pressure and temperature relevant to geological carbon storage. The simulations show
that the degree of swelling caused by intercalation of CO2 strongly depends on the
initial water content in the interlayer space and that CO2 intercalation stimulates
inner-sphere adsorption of the positively charged interlayer ions on the internal clay
surfaces, which modifies the wetting properties of the surfaces. DFT-based molecular
dynamics simulations were used to interpret the origin of the observed shift in the
asymmetric stretch vibration of CO2 trapped in montmorillonite. The origin of the shift
is attributed to the electric field effects on the CO2 molecules induced by the water
molecules.
Authors Ya Zhou, Wissam A. Saidi, and Kristen A. Fichthorn
We investigate the origins of experimentally observed differences in the
structure-directing capabilities of polyvinylpyrrolidone (PVP) and polyethylene oxide
(PEO) for the shape-selective, colloidal synthesis of {100}-faceted Ag nanostructures.
Using dispersion-corrected density-functional theory, we calculate the binding energies
of polymer repeat units to Ag(100) and Ag(111). At the level of the repeat unit, the
energetic preference for PEO to bind to {100} facets (?40 meV) is half that of PVP. At
the chain level, we use the Kuhn length to define the polymer binding unit and this is 9
(3) repeat units for PVP (PEO). A thermodynamic model predicts that the {100} binding
selectivity of PVP is 107 times higher than that of PEO at synthesis temperatures. These
results are consistent with experiment and demonstrate that a distinguishing
characteristic of a successful polymer structure-directing agent is the pairing of
facet-selective binding of the repeat unit with a sufficiently stiff chain.
Authors Lynn Mandeltort, De-Li Chen, Wissam A. Saidi, J. Karl
Johnson, Milton W. Cole, and John T. Yates Jr.
Single-walled carbon nanotubes (SWNTs) exhibit high surface areas and precisely
defined pores, making them potentially useful materials for gas adsorption and
purification. A thorough understanding of the interactions between adsorbates and SWNTs
is therefore critical to predicting adsorption isotherms and selectivities. Metallic
(M-) and semiconducting (S-) SWNTs have extremely different polarizabilities that might
be expected to significantly affect the adsorption energies of molecules. We
experimentally and theoretically show that this expectation is contradicted, for both a
long chain molecule (n-heptane) and atoms (Ar, Kr, and Xe). Temperature-programmed
desorption experiments are combined with van der Waals corrected density functional
theory, examining adsorption on interior and exterior sites of the SWNTs. Our
calculations show a clear dependence of the adsorption energy on nanotube diameter but
not on whether the tubes are conducting or insulating. We find no significant
experimental or theoretical difference in adsorption energies for molecules adsorbed on
M- and S-SWNTs having the same diameter. Hence, we conclude that the differences in
polarizabilities between M- and S-SWNTs have a negligible influence on gas adsorption
for spherical molecules as well as for highly anisotropic molecules such as n-heptane.
We expect this conclusion to apply to all types of adsorbed molecules where van der
Waals interactions govern the molecular interaction with the SWNT.
Carboxylation of carbon nanotubes (CNTs) is a byproduct of acid oxidation treatments
that are applied routinely for several purposes including cleaning of CNTs and as a
first step of functionalization procedures. In this study, we employ density functional
calculations to study the atomic and electronic structures of side-wall
—COOH-functionalized zigzag CNTs and elucidate their dependence on the tube diameter
and the metallic or semiconducting character. Adsorption of a —COOH group shows a
covalent bonding character associated with a small charge transfer from the CNT to the
carboxyl groups. The amount of charge transfer, as well as the binding energy, of the
carboxyl to the CNT decreases with the tube diameter. We find that it is
thermodynamically more favorable for —COOH to adsorb in pairs on top of two
neighboring carbon atoms that are bonded along the tube axis. This clustering effect
becomes more favorable for larger diameter CNTs, because the difference in adsorption
energy between isolated and pair carboxylation increases with tube diameter.
Furthermore, we find that pair adsorption is not kinetically hindered and shows similar
activation energies to that of the isolated adsorption. The electronic mechanism for the
clustering effect is discussed.
Authors De-Li Chen, Lynn Mandeltort, Wissam A. Saidi, John T.
Yates, Jr., Milton W. Cole, and J. Karl Johnson
The differences in the polarizabilities of metallic (M) and semiconducting (S)
single-walled carbon nanotubes (SWNTs) might give rise to differences in adsorption
potentials. We show from experiments and van der Waals—corrected density functional
theory that the binding energies of Xe adsorbed on M- and S-SWNTs are nearly identical.
Temperature programed desorption experiments of Xe on purified M- and S-SWNTs give
similar peak temperatures, indicating that desorption kinetics and binding energies are
independent of the type of SWNT. Binding energies computed from vdW-corrected density
functional theory are in good agreement with experiments.
Authors Joanna Kauczor, Patrick Norman and Wissam A. Saidi
We present frequency-dependent polarizabilities and C 6 dipole-dipole dispersion
coefficients for a wide range of fullerene molecules including C60, C70, C78, C80, C82,
and C84. The static and dynamic polarizabilities at imaginary frequencies are computed
using time-dependent Hartree-Fock, B3LYP, and CAM-B3LYP ab initio methods by employing
the complex linear polarization propagator and are subsequently utilized to determine
the C 6 coefficients using the Casimir-Polder relation. Overall, the C60 and C70 average
static polarizabilities ???(0) agree to better than 2% with linear-response
coupled-cluster single double and experimental benchmark results, and the C 6
coefficient of C60 agrees to better than 1% with the best accepted value. B3LYP provides
the best agreement with benchmark results with deviations less than 0.1% in ???(0) and C
6. We find that the static polarizabilities and the C 6 coefficients are non-additive,
and scale, respectively, as N 1.2 and N 2.2 with the number of carbon atoms in the
fullerene molecule. The exponent for C 6 power-dependence on N is much smaller than the
value predicted recently based on a classical-metallic spherical-shell approximation of
the fullerenes.
Authors David A. Egger, Victor G. Ruiz, Wissam A. Saidi, Tomas
Bucko, Alexandre Tkatchenko, and Egbert Zojer
For organic and hybrid electronic devices, the physicochemical properties of the
contained interfaces play a dominant role. To disentangle the various interactions
occurring at such heterointerfaces, we here model a complex, yet prototypical,
three-component system consisting of a Cu–phthalocyanine (CuPc) film on a
3,4,9,10-perylene-tetracarboxylic-dianhydride (PTCDA) monolayer adsorbed on Ag(111). The
two encountered interfaces are similar, as in both cases there would be no bonding
without van der Waals interactions. Still, they are also distinctly different, as only
at the Ag(111)–PTCDA interface do massive charge-rearrangements occur. Using recently
developed theoretical tools, we show that it has become possible to provide atomistic
insight into the physical and chemical processes in this comparatively complex
nanostructure distinguishing between interactions involving local rearrangements of the
charge density and long-range van der Waals attraction.
Authors Wissam A. Saidi, Haijun Feng, and Kristen A. Fichthorn
We use dispersion-corrected density functional theory (DFT) to resolve the role of
polyvinylpyrrolidone (PVP) in the shape-selective synthesis of Ag nanostructures by
probing the interaction of its 2-pyrrolidone (2P) ring with Ag(100) and Ag(111). We
employ two different semiempirical methods for including van der Waals (vdW)
interactions in DFT calculations: DFT+vdWsurf and DFT-D2. We find that DFT-D2, in its
original parametrization, overestimates the Ag metal dispersion interaction and causes
an unphysical herringbone-like reconstruction of Ag(100). This can be remedied in DFT-D2
by using modified vdW parameters for Ag that account for many-body screening effects.
The results obtained using DFT-D2 with the modified parameters agree well with
experiment and with DFT+vdWsurf results. We find that 2P binds more strongly to Ag(100)
than Ag(111), consistent with experiment. We analyze the origins of the
surface-sensitive binding and find that vdW attraction is stronger on Ag(111), but the
direct chemical bonding of 2P is stronger on Ag(100). We also study the influence of
strain on binding energies and find that tension tends to lower the vdW interaction with
the surfaces, while increasing the direct chemical-bonding interaction, consistent with
the d-band center model. Overall, our work indicates that strain has little impact on
the structure-directing capabilities of PVP, which is consistent with the fact that
strained, 5-fold twinned Ag nanowires have extensive {100} facets and relative small
{111} facets.
Authors Guangwen Zhou, Langli Luo, Liang Li, Jim Ciston, Eric A.
Stach, Wissam A. Saidi and Judith C. Yang
Oxidation of Cu occurs via Cu2O islanding on an oxide wetting layer at a critical
thickness of two atomic layers. The transition from 2D wetting-layer growth to 3D oxide
islanding is driven energetically arising from the Cu–Cu2O interfacial
interaction.
Authors Wissam A. Saidi, Minyoung Lee, Liang Li, Guangwen Zhou,
and Alan J. H. McGaughey
Using an ab initio atomistic thermodynamics framework, we identify the
stable surface structures during the early stages of Cu(100) oxidation at finite
temperature and pressure conditions. We predict the clean surface, the 0.25 monolayer
oxygen-covered surface, and the missing-row reconstruction as thermodynamically stable
structures in range of 100–1000 K and 10?15–105atm, consistent with previous
experimental and theoretical results. We also investigate the thermodynamic stabilities
of possible precursors to Cu2O formation including missing-row reconstruction structures
that include extra on- or subsurface oxygen atoms as well as boundary phases formed from
two missing-row nanodomains. While these structures are not predicted to be
thermodynamically stable for oxygen chemical potentials below the nucleation limit of
Cu2O, they are likely to exist due to kinetic hindrance.
Authors John Mark P. Martirez, Erie H. Morales, Wissam A. Saidi,
Dawn A. Bonnell, and Andrew M. Rappe
This contribution presents a study of the atomic and electronic structure of
the (5?×5?)R26.6°surface reconstruction
on BaTiO3 (001) formed by annealing in ultrahigh vacuum at
1300 K. Through density functional theory calculations in concert with
thermodynamic analysis, we assess the stability of
several BaTiO3 surface reconstructions and construct a phase
diagram as a function of the chemical potential of the constituent elements. Using both
experimental scanning tunneling microscopy (STM) and scanning tunneling spectroscopy
measurements, we were able to further narrow down the candidate structures, and conclude
that the surface is either TiO2-Ti3/5, TiO2-Ti4/5, or some combination, where
Ti adatoms occupy hollow sites of the TiO2 surface. Density functional theory
indicates that the defect states close to the valence band are from Ti
adatom 3d orbitals (?1.4??eV below the conduction band edge) in agreement
with scanning tunneling spectroscopy measurements showing defect
states 1.56±0.11??eV below the conduction band minimum
(1.03±0.09??eV below the Fermi level). STM measurements show electronic
contrast between empty and filled states’ images. The calculated local density of
states at the surface shows that Ti 3d states below and above the Fermi level
explain the difference in electronic contrast in the experimental STM images by the
presence of electronically distinctive arrangements of Ti adatoms. This work provides an
interesting contrast with the related oxide SrTiO3, for which the (001)
surface (5?×5?)R26.6° reconstruction is reported to be
theTiO2 surface with Sr adatoms.
Authors De-Li Chen, W A Al-Saidi and J Karl Johnson
Adsorption of noble gases on metal surfaces is determined by weak interactions. We
applied two versions of the nonlocal van der Waals density functional (vdW-DF) to
compute adsorption energies of Ar, Kr, and Xe on Pt(111), Pd(111), Cu(111), and Cu(110)
metal surfaces. We compared our results with data obtained using other density
functional approaches, including the semiempirical vdW-corrected DFT-D2. The vdW-DF
results show considerable improvements in the description of adsorption energies and
equilibrium distances over other DFT based methods, giving good agreement with
experiments. We also calculated perpendicular vibrational energies for noble gases on
the metal surfaces using vdW-DF data and found excellent agreement with available
experimental results. Our vdW-DF calculations show that adsorption of noble gases on
low-coordination sites is energetically favored over high-coordination sites, but only
by a few meV. Analysis of the two-dimensional potential energy surface shows that the
high-coordination sites are local maxima on the two-dimensional potential energy surface
and therefore unlikely to be observed in experiments; this provides an explanation of
the experimental observations. The DFT-D2 approach with the standard parameterization
was found to overestimate the dispersion interactions, and to give the wrong adsorption
site preference for four of the nine systems we studied.
Authors Dan C. Sorescu, Junseok Lee, Wissam A.
Al-Saidi and Kenneth D. Jordan
Adsorption and reactions of CO2 in the presence of H2O and OH species on the
TiO2 rutile (110)-(1×1) surface were investigated using dispersion-corrected
density functional theory and scanning tunneling microscopy. The coadsorbed H2O (OH)
species slightly increase the CO2adsorption energies, primarily through formation of
hydrogen bonds, and create new binding configurations that are not present on the
anhydrous surface.Proton transferreactions to CO2 with formation of bicarbonate and
carbonic acid species were investigated and found to have barriers in the range
6.1–12.8 kcal/mol, with reactions involving participation of two or more water
molecules or OH groups having lower barriers than reactions involving a single adsorbed
water molecule or OH group. The reactions to form the most stable adsorbed formate and
bicarbonate species are exothermic relative to the unreacted adsorbed CO2 and H2O
(OH) species, with formation of the bicarbonate species being favored. These results are
consistent with single crystal measurements which have identified formation of
bicarbonate-type species following coadsorption of CO2 and water on rutile
(110).
BNB is a challenging example of artifactual symmetry-breaking effects due to its
susceptibility to a pseudo second-order Jahn–Teller interaction, which results in a
structure with unequal BN bondlengths. The fixed-node diffusion Monte Carlo method is
employed to calculate the potential curves along the symmetric and asymmetric stretching
coordinates. With a multi-determinant wavefunction, the symmetric and asymmetric
structures are found degenerate within statistical errors, with the asymmetric
configuration lower in energy. The energy difference between the two structures is
smaller than the multi-reference coupled cluster result obtained with four determinants,
and supports previous conclusions that BNB has a floppy quasi-symmetric
ground-state.
Authors Guozhen Zhang, W. A. Al-Saidi, Evgeniy M.
Myshakin, and Kenneth D. Jordan
Water adsorption on the (001) surface of pyrophyllite [Al(OH)(Si2O5)] was
investigated using density functional theory (DFT) with dispersion corrections and force
field calculations. The DFT calculations show that a water molecule can bind either to
one or to two basal oxygen atoms of the surface, with adsorption energies varying from
?0.10 to ?0.19 eV depending on the binding configuration and binding site. Because the
water–water interactions are stronger than the water–surface interactions, the
energetically preferred structures with two or more molecules on the surface are
clusters reminiscent of their gas-phase counterparts. The trend in water–surface
binding energies with the number of water molecules obtained from force field
calculations qualitatively agrees with that predicted by the dispersion-corrected DFT
calculations. However, the force field calculations give a low-energy structural motif
with a water molecule coordinated to a hydroxyl group associated with the octahedral
layer of the pyrophyllite surface. This binding motif is found to be unstable in the DFT
calculations.
Authors W. A. Al-Saidi, Sanford A. Asher, and Patrick Norman
Geometries, UV absorption bands, and resonance Raman (RR) cross sections of TNT and
RDX are investigated using density functional theory (DFT) in conjunction with the
Coulomb attenuated B3LYP exchange-correlation functional. The absorption and RR spectra
are determined with use of vibronic (VB) theory, excited-state gradient, and complex
polarizability (CPP) approximations. We examined low-energy isomers (two for TNT and
four for RDX) whose energies differ by less than 1 kcal/mol, such that they would
appreciably be populated at room temperature. The two TNT isomers differ by an internal
rotation of the methyl group, while the four conformers of RDX differ by the
arrangements of the nitro group relative to the ring. Our theoretical optical properties
of the TNT and RDX isomers are in excellent agreement with experimental and recent
CCSD-EOM results, respectively. For the two TNT isomers, the ultraviolet RR (UVRR)
spectra are similar and in good agreement with recently measured experimental results.
Additionally, the UVRR spectra computed using the excited-state and CPP approaches
compare favorably with the VB theory results. On the other hand, the RR spectra of the
RDX conformers differ from one another, reflecting the importance of the positioning of
the NO2 groups with respect to the ring. In the gas phase or in solution, RDX would
give a spectrum associated with a conformationally averaged structure. It is encouraging
that the computed spectra of the conformers show similarities to recent measured RDX
spectra in acetonitrile solution, and reproduce the 10-fold decrease in the absolute
Raman cross sections of RDX compared to TNT for the observed 229 nm excitation. We show
that in TNT and RDX vibrational bands that couple to NO2 or the ring are
particularly resonance enhanced. Finally, the computed RDX spectra of the conformers
present a benchmark for understanding the RR spectra of the solid-phase polymorphs of
RDX.
Authors W. A. Al-Saidi, Vamsee K. Voora, and Kenneth D.
Jordan
The Tkatchenko–Scheffler vdW-TS method [Phys. Rev. Lett.2009,160;102, 073005] has
been implemented in a plane-wave DFT code and used to characterize several
dispersion-dominated systems, including layered materials, noble-gas solids, and
molecular crystals. Full optimizations of the structures, including relaxation of the
stresses on the unit cells, were carried out. Internal geometrical parameters, lattice
constants, bulk moduli, and cohesive energies are reported and compared to experimental
results.
Authors W. A. Al-Saidi, Haijun Feng, and Kristen A.
Fichthorn
We use density functional theory to resolve the role of polyvinylpyrrolidone (PVP)
in the shape-selective synthesis of Ag nanostructures. At the segment level, PVP binds
more strongly to Ag(100) than Ag(111) because of a surface-sensitive balance between
direct binding and van der Waals attraction. At the chain level, correlated segment
binding leads to a strong preference for PVP bind to Ag(100). Our study underscores
differences between small-molecule and polymeric structure-directing agents.
Authors De-Li Chen, W. A. Al-Saidi, and J. Karl Johnson
We use a nonlocal van der Waals density functional (vdW-DF) approach to reexamine
the problem of why noble gases are experimentally observed to adsorb on low-coordination
atop sites rather than on high-coordination hollow sites for several different metal
surfaces. Previous calculations using density functional theory (DFT) within the local
density approximation (LDA) ascribed the site preference to reduced Pauli repulsion at
atop sites, largely due to reduced exchange repulsion within LDA-DFT. In contrast, our
vdW-DF calculations show that site preference is not due to differences in the exchange
repulsion at all, but rather the result of a delicate balance between the electrostatic
and kinetic energies; surprisingly, exchange-correlation energies play a negligible role
in determining site preference. In contrast to previous calculations, we find that
experimental results cannot be explained in terms of binding energy differences between
atop and hollow sites. Instead, we show that the hollow sites are transition states
rather than minima on the two-dimensional potential energy surface, and therefore not
likely to be observed in experiments. This phenomenon is quite general, holding for
close-packed and non-close-packed metal surfaces. We show that inclusion of nonlocal vdW
interactions is crucial for obtaining results in quantitative agreement with experiments
for adsorption energies, equilibrium distances, and vibrational energies.
Authors Dan C. Sorescu, Wissam A.
Al-Saidi and Kenneth D. Jordan
Adsorption,diffusion, and dissociation of CO2 on the anatase (101) surface were
investigated using dispersion-corrected density functional theory. On the
oxidizedsurface several different local minima were identified of which the most stable
corresponds to a CO2 molecule adsorbed at a five-fold coordinated Ti site in a
tilted configuration. Surfacediffusion is characterized by relatively small activation
barriers. Preferential diffusion takes place along Ti rows and involves a cartwheel type
of motion. The presence of a bridging oxygen defect or a surfaceinterstitial Ti atom
allows creation of several new strong binding configurations the most stable of which
have bent CO2 structures with simultaneous bonding to two surface Ti atoms.
Subsurface oxygen vacancy or interstitial Ti defects are found to enhance the bonding of
CO2 molecules to the surface. CO2dissociation from these defect sites is calculated
to be exothermic with barriers less than 21 kcal/mol. The use of such defects for
catalytic activation of CO2 on anatase (101) surface would require a mechanism for
their regeneration.
Authors Amy M. Beaird,Ping Li,Hilary S. Marsh,W. A. Al-Saidi,J.
Karl Johnson,Michael A. Matthews, and Christopher T. Williams
Sodium metaborate hydrates are a class of compounds represented by the stoichiometry
NaBO2xH2O. Recently, sodium metaborate has received attention as the byproduct of sodium
borohydride hydrolysis, a reaction that is under consideration for hydrogen storage
applications. The aim of this work was to understand the disposition of water in the
crystal structure of hydrated sodium metaborates and to characterize the thermal
stability and dehydration of the various hydrated species to optimize hydrogen storage
efficiency as well as recyclability of the borate. Observations from a suite of
analytical techniques including thermal analyses (thermogravimetric
analysis/differential scanning calorimetry), X-ray diffraction, and Raman spectroscopy
were correlated to characterize the dehydration mechanism of commercially available
sodium metaborates, with an emphasis on the dihydrate (x = 2). A transformation
from tetrahedrally coordinated boron to trigonal boron occurs when
NaB(OH)4 (x = 2) is heated between 25 and 400 °C. The first dehydration
to Na3[B3O5(OH)2] (x = 1/3) releases 5 mol of water between 83 and 155
°C. The final mole of water is released between 249 and 280 °C, and
Na3B3O6 (x = 0) is formed. Raman spectra are reported for x = 2
and 1/3 for the first time. First-principles density functional theory was
used to compute Raman spectra of the x = 1/3 and 2 material in order
to assign the modes. We found reasonably good agreement between the experimentally
measured and calculated vibrational frequencies.
Authors Vamsee K. Voora, W. A. Al-Saidi, andKenneth D. Jordan
The stacking parameters, lattice constants, bond lengths, and bulk moduli of
pyrophyllite and montmorillonites (MMTs), with alkali and alkali earth metal ions, are
investigated using density functional theory with and without dispersion corrections.
For pyrophyllite, it is found that the inclusion of the dispersion corrections
significantly improves the agreement of the calculated values of the lattice parameters
and bulk modulus with the experimental values. For the MMTs, the calculations predict
that the interlayer spacing varies approximately linearly with the cation radius. The
inclusion of dispersion corrections leads to sizable shifts of the interlayer spacings
to shorter values. In Li-MMT, compaction of the interlayer distance triggers migration
of the Li ion into the tetrahedral sheet and close coordination with basal oxygen atoms.
Analysis of electron density distributions shows that the isomorphic octahedral
Al3+/Mg2+ substitution in MMT causes an increase of electron density on the basal
oxygen atoms of the tetrahedral sheets.
Authors Glen R. Jenness, Ozan Karalti, W. A. Al-Saidi,
and Kenneth D. Jordan
The interaction of a water monomer with a series of linear acenes (benzene,
anthracene, pentacene, heptacene, and nonacene) is investigated using a wide range of
electronic structure methods, including several “dispersionâ€-corrected density
functional theory (DFT) methods, several variants of the random phase approximation
(RPA), DFT-based symmetry-adapted perturbation theory with density fitting
(DF-DFT-SAPT), MP2, and coupled-cluster methods. The DF-DFT-SAPT calculations are used
to monitor the evolution of the electrostatics, exchange-repulsion, induction, and
dispersion contributions to the interaction energies with increasing acene size and also
provide the benchmark data against which the other methods are assessed.
Authors Dan C. Sorescu, Junseok Lee, Wissam A.
Al-Saidi and Kenneth D. Jordan
Adsorption of CO2 on the rutile(110) surface was investigated using
dispersion-corrected density functional theory and scanning tunneling microscopy(STM).
On the oxidizedsurface the CO2 molecules are found to bind most strongly at the
five-fold coordinated Ti sites adopting tilted or flat configurations. The presence of
bridging oxygen defects introduces two new adsorption structures, the most stable of
which involves CO2 molecules bound in tilted configurations at the defect sites.
Inclusion of dispersion corrections in the density functional theory calculations leads
to large increases in the calculated adsorptionenergies bringing these quantities into
good agreement with experimental data. The STM measurements confirm two of the
calculated adsorption configurations.
We present an ab initio density functional study of ferroelectricity in
single-domain PbTiO3-based nanocapacitors. We used density functional theory
with the recently introduced PBEsol generalized-gradient exchange-correlation
functional, which we found to give accurate properties of bulk ferroelectric (FE)
materials. Pt and Au electrodes are used in our study to gain a thorough understanding
of the electrode-oxide interfaces, and the role of the interfacial chemical bonding and
charge transfer in stabilizing the FE polar phase. We found that the FE properties of
the thin films depend not only on the electrode and the FE material but also on the
electrode-perovskite termination (TiO2 vs PbO), exemplifying the key role of the
interface in these systems. The critical thickness was found to be 24–28?Å.
In addition, a Löwdin orbital analysis gives a detailed description of the
distribution of charges in the system, and shows the importance of charge passivation by
the electrodes in stabilizing the FE polar phase.
Authors F.-F. Wang, G. Jenness, W. A.
Al-Saidi and K. D. Jordan
Localized molecular orbital energy decomposition analysis and symmetry-adapted
perturbation theory (SAPT) calculations are used to analyze the two- and three-body
interactionenergies of four low-energy isomers of (H2O)6 in order to gain
insight into the performance of several popular density functionals for describing the
electrostatic, exchange-repulsion, induction, and short-range dispersion interactions
between water molecules. The energy decomposition analyses indicate that all density
functionals considered significantly overestimate the contributions of charge transfer
to the interactionenergies. Moreover, in contrast to some studies that state that
density functional theory(DFT) does not include dispersion interactions, we adopt a
broader definition and conclude that for (H2O)6 the short-range dispersion
interactions recovered in the DFT calculations account about 75% or more of the net
(short-range plus long-range) dispersion energies obtained from the SAPT
calculations.
2009 and before
We present ab initio calculations of atomic and molecular systems containing
the first-, second-, and third-row post-d elements (Ga–Br, In–I, and Tl–At) using
several methods including variational and diffusion Monte Carlo. In the quantum Monte Carlo
calculations, we used the recent scalar-relativistic energy-consistent Hartree–Fock
pseudopotentials [M. Burkatzki et al., J. Chem. Phys.126, 234105 (2007)], which are
nonsingular at the origin. For the first- and second-row elements, the calculated
ionizationenergies and electron affinities are in excellent agreement with those obtained
using CCSD(T) with large basis sets and with experiment after correcting approximately for
spin-orbit effects. For the third-row elements, where relativistic effects cannot be
adequately included by a simple j-averaging, the results are in excellent agreement
with CCSD(T) energies obtained with a large (5-zeta) basis set. Benchmark calculations of
the dissociation energies, vibration frequencies, and equilibrium bond lengths of several
diatomic molecules including As2, Br2, Sb2, and I2 as well as the
hydrides XH (X=Ga, Br, In, I, and At) are presented.
Diffusion Monte Carlo (DMC) calculations are performed on the monocyclic and bicyclic
forms ofm-benzyne, which are the equilibrium structures at the CCSD(T) and CCSD levels of
coupled clustertheory. We employed multiconfiguration self-consistent field trial wave
functions which are constructed from a carefully selected eight-electrons-in-eight-orbitals
complete active space [CAS(8,8)], with configuration state function coefficients that are
reoptimized in the presence of a Jastrow factor. The DMC calculations show that the
monocyclic structure is lower in energy than the bicyclic structure by 1.9(2)kcal?mole,
which is in excellent agreement with the best coupled cluster results.
Authors Wirawan Purwanto, W. A. Al-Saidi, Henry
Krakauer and Shiwei Zhang
The use of an approximate reference state wave function in electronic many-body methods
can break the spin symmetry of Born-Oppenheimer spin-independent Hamiltonians. This can
result in significant errors, especially when bonds are stretched or broken. A simple
spin-projection method is introduced for auxiliary-field quantum Monte Carlo (AFQMC)
calculations, which yields spin-contamination-free results, even with a
spin-contaminated ??r?. The method is applied to the difficult F2 molecule,
which is unbound within unrestricted Hartree–Fock (UHF). With a UHF ??r?, spin
contamination causes large systematic errors and long equilibration times in AFQMC in the
intermediate, bond-breaking region. The spin-projection method eliminates these problems and
delivers an accurate potential energy curve from equilibrium to the dissociation limit using
the UHF ??r?. Realistic potential energy curves are obtained with a cc-pVQZ basis. The
calculated spectroscopic constants are in excellent agreement with experiment.
Authors W. A. Al-Saidi, E. J. Walter, and A. M. Rappe
We report Hartree-Fock (HF)-based pseudopotentials suitable for plane-wave calculations.
Unlike typical effective core potentials, the present pseudopotentials are finite at the
origin and exhibit rapid convergence in a plane-wave basis; the optimized pseudopotential
method [A. M. Rappe et al., Phys. Rev. B 41, 1227 (1990)] improves plane-wave
convergence. Norm-conserving HF pseudopotentials are found to develop long-range
non-Coulombic behavior which does not decay faster than 1?r, and is nonlocal. This
behavior, which stems from the nonlocality of the exchange potential, is remedied using a
recently developed self-consistent procedure [J. R. Trail and R. J. Needs, J. Chem.
Phys. 122, 014112 (2005)]. The resulting pseudopotentials slightly violate the norm
conservation of the core charge. We calculated several atomic properties using these
pseudopotentials, and the results are in good agreement with all-electron HF values. The
dissociation energies, equilibrium bond lengths, and frequencies of vibration of several
dimers obtained with these HF pseudopotentials and plane waves are also in good agreement
with all-electron results.
Authors W. A. Al-Saidi, Shiwei Zhang and Henry Krakauer
Bond stretching mimics different levels of electron correlation and provides a
challenging test bed for approximate many-body computational methods. Using the recently
developed phaseless auxiliary-field quantum Monte Carlo (AF QMC) method, we examine bond
stretching in the well-studied molecules BH and N2 and in the H50 chain. To control the
sign/phase problem, the phaseless AF QMC method constrains the paths in the auxiliary-field
path integrals with an approximate phase condition that depends on a trial wave function.
With single Slater determinants from unrestricted Hartree-Fock as trial wave function, the
phaseless AF QMC method generally gives better overall accuracy and a more uniform behavior
than the coupled cluster CCSD(T) method in mapping the potential-energy curve. In both BH
and N2, we also study the use of multiple-determinant trial wave functions from
multiconfiguration self-consistent-field calculations. The increase in computational cost
versus the gain in statistical and systematic accuracy are examined. With such trial wave
functions, excellent results are obtained across the entire region between equilibrium and
the dissociation limit.
Authors W. A. Al-Saidi, Henry Krakauer and Shiwei Zhang
The authors present phaseless auxiliary-field (AF) quantum Monte Carlo (QMC)
calculations of the ground states of some hydrogen-bonded systems. These systems were
selected to test and benchmark different aspects of the new phaseless AF QMC method. They
include the transition state of H+H2 near the equilibrium geometry and in the van der Walls
limit, as well as the H2O, OH, and H2O2 molecules. Most of these systems present significant
challenges for traditional independent-particle electronic structure approaches, and many
also have exact results available. The phaseless AF QMC method is used either with a plane
wave basis with pseudopotentials or with all-electron Gaussian basis sets. For some systems,
calculations are done with both to compare and characterize the performance of AF QMC under
different basis sets and different Hubbard-Stratonovich decompositions. Excellent results
are obtained using as input single Slater determinant wave functions taken from
independent-particle calculations. Comparisons of the Gaussian based AF QMC results with
exact full configuration interaction show that the errors from controlling the phase problem
with the phaseless approximation are small. At the large basis-size limit, the AF QMC
results using both types of basis sets are in good agreement with each other and with
experimental values.
Authors W. A. Al-Saidi, Henry Krakauer and Shiwei Zhang
A series of calculations for the first- and second-row post-d elements (Ga--Br and
In--I) are presented using the phaseless auxiliary-field quantum Monte Carlo (AF QMC)
method. This method is formulated in a Hilbert space defined by any chosen one-particle
basis and maps the many-body problem into a linear combination of independent-particle
solutions with external auxiliary fields. The phase/sign problem is handled approximately by
the phaseless formalism using a trial wave function, which in our calculations was chosen to
be the Hartree-Fock solution. We used the consistent correlated basis sets of Peterson et
al. [J. Chem. Phys.119, 11099 (2003);119, 11113 (2003)], which employ a small-core
relativistic pseudopotential. The AF QMC results are compared with experiment and with those
from density functional (generalized gradient approximation and B3LYP) and CCSD(T)
calculations. The AF QMC total energies agree with CCSD(T) to within a few millihartrees
across the systems and over several basis sets. The calculated atomic electron
affinities,ionization energies, and spectroscopic properties of dimers are, at large basis
sets, in excellent agreement with experiment.
Authors W. A. Al-Saidi, Shiwei Zhang and Henry Krakauer
We extend the recently introduced phaseless auxiliary-field quantum Monte Carlo (QMC)
approach to any single-particle basis and apply it to molecular systems with Gaussian basis
sets. QMC methods in general scale favorably with the system size as a low power. A QMC
approach with auxiliary fields, in principle, allows an exact solution of the
Schrödinger equation in the chosen basis. However, the well-known sign/phase problem
causes the statistical noise to increase exponentially. The phaseless method controls this
problem by constraining the paths in the auxiliary-field path integrals with an approximate
phase condition that depends on a trial wave function. In the present calculations, the
trial wave function is a single Slater determinant from a Hartree-Fock calculation. The
calculated all-electron total energies show typical systematic errors of no more than a few
millihartrees compared to exact results. At equilibrium geometries in the molecules we
studied, this accuracy is roughly comparable to that of coupled cluster with single and
double excitations and with noniterative triples [CCSD(T)]. For stretched bonds in H2O, our
method exhibits a better overall accuracy and a more uniform behavior than CCSD(T).
Authors W. A. Al-Saidi, Henry Krakauer, and Shiwei Zhang
Calculations of the binding energy of the transition-metal oxide molecules TiO and MnO
are presented, using a recently developed phaseless auxiliary-field quantum Monte Carlo
approach. This method maps the interacting many-body problem onto a linear combination of
noninteracting problems by a complex Hubbard-Stratonovich transformation, and controls the
phase and sign problem with a phaseless approximation relying on a trial wave function. It
employs random walks in Slater determinant space to project the ground state of the system,
and allows use of much of the same machinery as in standard density functional theory
calculations using the plane-wave basis and nonlocal pseudopotentials. The calculations used
a single Slater determinant trial wave function obtained from a density functional
calculation, with no further optimization. The calculated binding energies are in good
agreement with experiment and with recent diffusion Monte Carlo results. Together with
previous results for sp-bonded systems, the present study indicates that the phaseless
auxiliary-field method is a robust and promising approach for the study of correlation
effects in real materials.
Authors Shiwei Zhang, , Henry Krakauer, Wissam A. Al-Saidi, Malliga
Suewattana
To treat interacting quantum systems, it is often crucial to have accurate calculations
beyond the mean-field level. Many-body simulations based on field-theoretical approaches are
a promising tool for this purpose and are applied in several sub-fields of physics, in
closely related forms. An major difficulty is the sign or phase problem, which causes the
Monte Carlo variance to increase exponentially with system size. We address this issue in
the context of auxiliary-field simulations of realistic electronic systems in condensed
matter physics. We show how to use importance sampling of the complex fields to control the
phase problem. An approximate approach is formulated with a trial determinant to constrain
the paths in field space and completely eliminate the growth of the noise. For ab initio
electronic structure calculations, this gives a many-body approach in the form of a
“coherent†superposition of mean-field calculations, allowing direct incorporation of
state-of-the-art technology from the latter (non-local pseudopotentials; high quality basis
sets, etc.). In our test calculations, single Slater determinants from density functional
theory or Hartree–Fock calculations were used as trial wave functions, with no additional
optimization. The calculated dissociation energies of various molecules and the cohesive
energy of bulk Si are in excellent agreement with experiment and are comparable to or better
than the best existing theoretical results.
We have studied the superconducting–insulating phase transition in a disordered
two-dimensional Josephson junction array, using quantum Monte Carlo techniques. We consider
disorder in both the capacitive energies and in the values of the offset charges. The
calculated phase diagram shows that the lobe structure of the phase diagram disappears for
sufficiently strong disorder in the offset charge. Our results agree quite well with the
previous calculations carried out using a mean-field approximation.
We have numerically investigated the dynamics of vortices in a clean layered
superconductor placed in a perpendicular magnetic field. We describe the energetics using a
Ginzburg-Landau free-energy functional in the lowest-Landau-level approximation. The
dynamics are determined using the time-dependent Ginzburg-Landau approximation, and thermal
fluctuations are incorporated via a Langevin term. The c-axis conductivity at nonzero
frequencies, as calculated from the Kubo formalism, shows a strong but not divergent
increase as the melting temperature TM is approached from above, followed by an apparently
discontinuous drop at the vortex-lattice freezing temperature. The discontinuity is
consistent with the occurrence of a first-order freezing. The calculated equilibrium
properties agree with previous Monte Carlo studies using the same Hamiltonian. We briefly
discuss the possibility of detecting this fluctuation conductivity experimentally.
We study the zero temperature (T=0) quantum rotor model with on-site disorder in the
charging energy. Such a model may serve as an idealized Hamiltonian for an array of
Josephson-coupled small superconducting grains or superfluid 4He in a disordered
environment. In the approximation of small-amplitude phase fluctuations, the Hamiltonian
maps onto a system of coupled harmonic oscillators with on-site disorder. We study the
effects of disorder in this harmonic regime, using the coherent potential approximation,
obtaining the density of states and the lifetimes of the spin-wave-like excitations for
several choices of the parameters which characterize the disorder. Finally, we estimate the
parameters characterizing the T=0 quantum melting of the phase order, using a suitable
Lindemann criterion.
We have studied quantum mechanically a system of several small identical Josephson
junctions in a lossless single-mode cavity for different initial states, under conditions
such that the system is at resonance. This system is analogous to a collection of identical
atoms in a cavity, which is described under appropriate conditions by the Dicke model. We
find that our system can be well approximated by a reduced Hamiltonian consisting of two
levels per junction. The reduced Hamiltonian is similar to the Dicke Hamiltonian, but
contains an additional term resembling a dipole-dipole interaction between the junctions.
This extra term can be understood as a natural consequence of degenerate second-order
(Löwdin) perturbation theory. For typical, physically reasonable values of the
junction-cavity coupling, we find that this perturbation treatment is an adequate way to
include the junction energy levels beyond the lowest two. As in the Dicke model, we find
that, when N junctions are present in the cavity, the junction-cavity interaction is
enhanced by N???, with a corresponding decrease in the Rabi oscillation period. We find that
this enhancement survives even if the junctions differ slightly from one another, as
expected in a realistic system. Since coherence effects thus reduce the Rabi period, it may
become smaller than the decoherence time due to dissipation, making these oscillations
observable.
We carry out a quantum-mechanical analysis of a small Josephson junction coupled to a
single-mode resonant cavity. We find that the eigenstates of the combined junction-cavity
system are strongly entangled only when the gate voltage applied at one of the
superconducting islands is tuned to certain special values. One such value corresponds to
the resonant absorption of a single photon by Cooper pairs in the junction. Another special
value corresponds to a two-photon absorption process. Near the single-photon resonant
absorption, the system is accurately described by a simplified model in which only the
lowest two levels of the Josephson junction are retained in the Hamiltonian matrix. We
noticed that this approximation does not work very well as the number of photons in the
resonator increases. Our system shows also the phenomenon of “collapse and revivalâ€
under suitable initial conditions, and our full numerical solution agrees with the two level
approximation result.