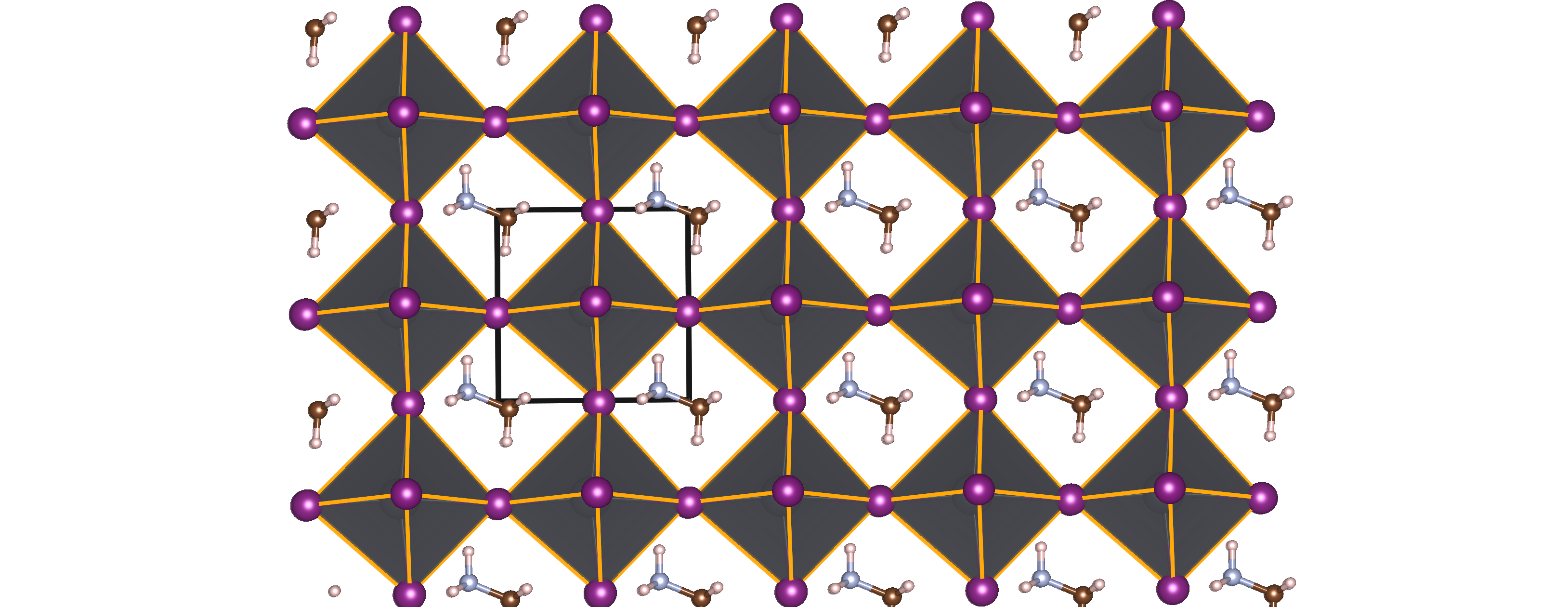
Solution-processable organo-metal halide perovskites (OMHPs) with a
solar conversion efficiency of nearly 20.1% are garnering increased
attention for third-generation photovoltaic technologies. The interest
in these materials started shortly after the first realization of
their potential photovoltaic properties, and their utilization in
low-cost solution-processable meso-superstructured solar cells. This
instigated many experimental and theoretical studies of the OMHPs all
emphasizing their exceptional properties for photovoltaic
applications. The most investigated OMHPs member is methylammonium
lead iodide MAPbI3 (MA+=CH3NH3+), which has the best performance as a
photovoltaic absorbent.
Author
Yaguang Guo, Qian Wang, and Wissam A. Saidi
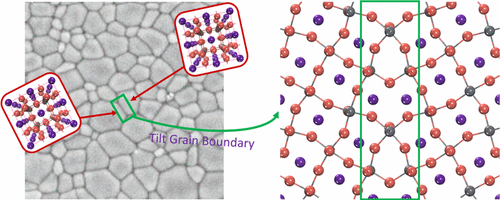
Organometal trihalide perovskites are emerging as very promising photovoltaic materials, which is rivaling that of single crystal silicon solar cells despite their polycrystalline nature with relatively high density of grain boundaries (GBs). There is a lack of understanding of the effects of GBs on halide perovskites as their presence in silicon and other photovoltaic materials is generally detrimental to their photovoltaic properties. Using first-principles calculations, we systematically investigate the geometric structures of high-angle tilt GBs in halide perovskites CsPbX3 (X = Cl, Br, and I) starting from the coincidence site lattice model and refining using crystal shifts and lattice expansion. Electronic density of states calculations reveal that GBs in halides perovskites do not generate midgap states because of the large distance between the unsaturated atoms and the atomic reconstructions in the GB region. However, we show that the GBs can induce different very shallow states near the valence band edge that can hinder hole diffusion. We further extend the results to MAPbI3 GBs and also show their benign effect on optoelectronic properties.
Author
Benjamin J. Foley, Justin Girard, Blaire A. Sorenson, Alexander Z. Chen, J. Scott Niezgoda, Matthew R. Alpert, Angela F. Harper, Detlef-M. Smilgies, Paulette Clancy, Wissam A. Saidi and Joshua J. Choi
Accelerating the progress toward realizing metal halide perovskite solar cells with improved efficiency, stability and reliability requires a deeper understanding of the thin film formation processes. This paper investigates the impact of rationally selected chemical additives in precursor solutions on the nucleation and growth of metal halide perovskite thin films. Computational screening was performed to guide the selection of tetrahydrothiophene oxide as an additive with stronger solvation efficacy than all other commonly used solvents. In situ grazing incidence X-ray diffraction measurements show that the additives suppress the formation of homogeneous nuclei as well as crystalline intermediate structures. Instead, heterogeneous nucleation on the substrate surface and growth of a thin film with a strongly preferential crystallographic orientation occur directly from the precursor solution. Density functional theory calculations show that the crystallographic orientation of the thin films can be tuned by altering the surface energies with the chemical additives. The crystallographic orientation of the thin films is found to have a significant impact on the open circuit voltage of solar cell devices, highlighting the importance of controlling the metal halide perovskite thin film orientation for improved solar cell efficiency.
Author
Wissam A. Saidi, Samuel Ponce, and Bartomeu Monserrat
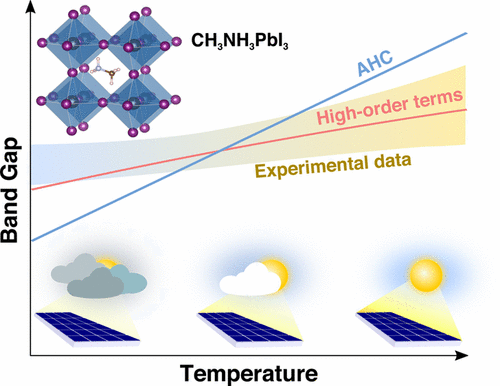
Environmental effects and intrinsic energy-loss processes lead to fluctuations in the operational temperature of solar cells, which can profoundly influence their power conversion efficiency. Here we determine from first-principles the effects of temperature on the band gap and band edges of the hybrid pervoskite CH3NH3PbI3 by accounting for electron–phonon coupling and thermal expansion. From 290 to 380 K, the computed band gap change of 40 meV coincides with the experimental change of 30–40 meV. The calculation of electron–phonon coupling in CH3NH3PbI3 is particularly intricate as the commonly used Allen–Heine–Cardona theory overestimates the band gap change with temperature, and excellent agreement with experiment is only obtained when including high-order terms in the electron–phonon interaction. We also find that spin–orbit coupling enhances the electron–phonon coupling strength but that the inclusion of nonlocal correlations using hybrid functionals has little effect. We reach similar conclusions in the metal–halide perovskite CsPbI3. Our results unambiguously confirm for the first time the importance of high-order terms in the electron–phonon coupling by direct comparison with experiment.
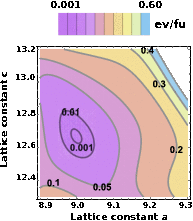
Hybrid organic-inorganic perovskites, as well as the perovskites in general, are known for their phase complexity evidenced by the stabilization of different polymorphs, and thus an understanding of their regions of stability and transitions can be important for their photovoltaic and optoelectronic technologies. Here we use a multiscale approach based on first-principles calculations with van der Waals corrections and classical force-field molecular dynamics to determine the finite-temperature properties of the tetragonal and cubic phases of CH3NH3PbI3. Temperature effects are implicitly included using the quasi-harmonic approximation that can describe anharmonic behavior due to thermal expansion through the dependence of the harmonic frequencies on structural parameters. Our finite-temperature free-energy surfaces predict the lattice and elastic moduli evolution with temperature, and show in particular that the calculated lattice parameters of the cubic and tetragonal phases are to within 1% of experimental values. Further, our results show that the phonons are the major contributing factor for stabilizing the cubic phase at high temperatures mainly due to the low-energy phonon modes that are associated with the inorganic lattice. On the other hand, the configurational entropy due to CH3NH3 + rotational degrees of freedom is slightly more favored in the cubic phase and amounts to less than 0.2% of the T = 0 K free-energy difference between the two phases.
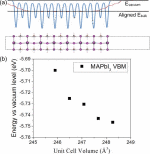
Author Benjamin J Foley, Daniel L Marlowe, Keye Sun, Wissam A Saidi, Louis Scudiero, Mool C Gupta, and Joshua J Choi
Temperature dependent energy levels of methylammonium lead iodide are investigated using a combination of ultraviolet photoemission spectroscopy and optical spectroscopy. Our results show that the valence band maximum and conduction band minimum shift down in energy by 110 meV and 77 meV as temperature increases from 28 °C to 85 °C. Density functional theory calculations using slab structures show that the decreased orbital splitting due to thermal expansion is a major contribution to the experimentally observed shift in energy levels. Our results have implications for solar cell performance under operating conditions with continued sunlight exposure and increased temperature.