Materials Science is largely concerned with how interfaces affect the properties of the heterostructures. Recent developments in experimental tools TEM and STM have provided an atomistic glimpse of the interface, which in conjunction with quantum mechanical simulations gave a comprehensive picture of the interface and its keys effects on the heterostructure.
Authors Wissam A. Saidi, John Mark P. Martirez, and Andrew M. Rappe
We present a systematic evaluation of the effects of polarization switchability on surface structure and stoichiometry in BaTiO3 and PbTiO3 ferroelectric oxides. We show that charge passivation, mostly by ionic surface reconstructions, is the driving force for the stability of the surfaces, which suggests that varying the substrate polarization offers a new mechanism for controlling surface reconstructions in polar systems and inducing highly nonstoichiometric structures. Conversely, for thin-films the chemical environment can drive polarization switching via induced compositional changes on the surface. We find that the value of the oxygen partial pressure for the positive-to-negative polar transition is in good agreement with the recent experimental value for thin-film PbTiO3. For BaTiO3, we show that it is harder for oxygen control to drive polar transition because it is more difficult to reduce. This study opens up the possibility of real-time control of structure and composition of oxide surfaces.
Authors Erie H. Morales, John Mark P. Martirez, Wissam A. Saidi, Andrew M. Rappe, and Dawn A. Bonnell
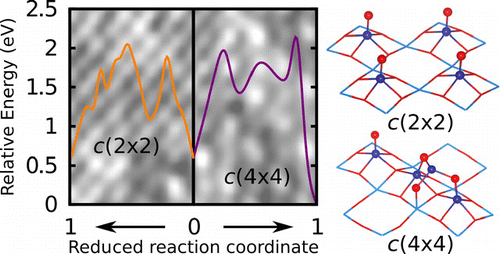
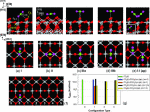
Coexistence of surface reconstructions is important due to the diversity in kinetic and thermodynamic processes involved. We identify the coexistence of kinetically accessible phases that are chemically identical and form coherent interfaces. Here, we establish the coexistence of two phases, c(2 × 2) and c(4 × 4), in BaTiO3(001) with atomically resolved Scanning Tunneling Microscopy (STM). First-principles thermodynamic calculations determine that TiO adunits and clusters compose the surfaces. We show that TiO diffusion results in a kinetically accessible c(2 × 2) phase, while TiO clustering results in a kinetically and thermodynamically stable c(4 × 4) phase. We explain the formation of domains based on the diffusion of TiO units. The diffusion direction determines the observed 1D coherent interfaces between c(2 × 2) and c(4 × 4) reconstructions. We propose atomic models for the c(2 × 2), c(4 × 4), and 1D interfaces.
Authors Wissam A. Saidi, Minyoung Lee, Liang Li, Guangwen Zhou, and Alan J. H. McGaughey
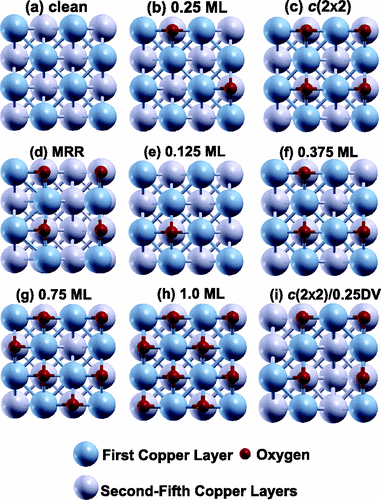
Using an ab initio atomistic thermodynamics framework, we identify the stable surface structures during the early stages of Cu(100) oxidation at finite temperature and pressure conditions. We predict the clean surface, the 0.25 monolayer oxygen-covered surface, and the missing-row reconstruction as thermodynamically stable structures in range of 100–1000 K and 10?15–105atm, consistent with previous experimental and theoretical results. We also investigate the thermodynamic stabilities of possible precursors to Cu2O formation including missing-row reconstruction structures that include extra on- or subsurface oxygen atoms as well as boundary phases formed from two missing-row nanodomains. While these structures are not predicted to be thermodynamically stable for oxygen chemical potentials below the nucleation limit of Cu2O, they are likely to exist due to kinetic hindrance.
Authors Dan C. Sorescu, Junseok Lee, Wissam A. Al-Saidi and Kenneth D. Jordan
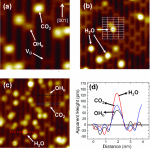
Adsorption and reactions of CO2 in the presence of H2O and OH species on the TiO2 rutile (110)-(1×1) surface were investigated using dispersion-corrected density functional theory and scanning tunneling microscopy. The coadsorbed H2O (OH) species slightly increase the CO2adsorption energies, primarily through formation of hydrogen bonds, and create new binding configurations that are not present on the anhydrous surface.Proton transferreactions to CO2 with formation of bicarbonate and carbonic acid species were investigated and found to have barriers in the range 6.1–12.8 kcal/mol, with reactions involving participation of two or more water molecules or OH groups having lower barriers than reactions involving a single adsorbed water molecule or OH group. The reactions to form the most stable adsorbed formate and bicarbonate species are exothermic relative to the unreacted adsorbed CO2 and H2O (OH) species, with formation of the bicarbonate species being favored. These results are consistent with single crystal measurements which have identified formation of bicarbonate-type species following coadsorption of CO2 and water on rutile (110).
Authors Dan C. Sorescu, Wissam A. Al-Saidi and Kenneth D. Jordan
Adsorption,diffusion, and dissociation of CO2 on the anatase (101) surface were investigated using dispersion-corrected density functional theory. On the oxidizedsurface several different local minima were identified of which the most stable corresponds to a CO2 molecule adsorbed at a five-fold coordinated Ti site in a tilted configuration. Surfacediffusion is characterized by relatively small activation barriers. Preferential diffusion takes place along Ti rows and involves a cartwheel type of motion. The presence of a bridging oxygen defect or a surfaceinterstitial Ti atom allows creation of several new strong binding configurations the most stable of which have bent CO2 structures with simultaneous bonding to two surface Ti atoms. Subsurface oxygen vacancy or interstitial Ti defects are found to enhance the bonding of CO2 molecules to the surface. CO2dissociation from these defect sites is calculated to be exothermic with barriers less than 21 kcal/mol. The use of such defects for catalytic activation of CO2 on anatase (101) surface would require a mechanism for their regeneration.
Authors Dan C. Sorescu, Junseok Lee, Wissam A. Al-Saidi and Kenneth D. Jordan
Adsorption of CO2 on the rutile(110) surface was investigated using dispersion-corrected density functional theory and scanning tunneling microscopy(STM). On the oxidizedsurface the CO2 molecules are found to bind most strongly at the five-fold coordinated Ti sites adopting tilted or flat configurations. The presence of bridging oxygen defects introduces two new adsorption structures, the most stable of which involves CO2 molecules bound in tilted configurations at the defect sites. Inclusion of dispersion corrections in the density functional theory calculations leads to large increases in the calculated adsorptionenergies bringing these quantities into good agreement with experimental data. The STM measurements confirm two of the calculated adsorption configurations.