Authors Lynn Mandeltort, De-Li Chen, Wissam A. Saidi, K. Johnson, Milton W. Cole, and J. T. Yates
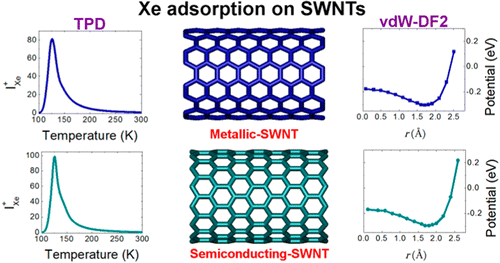
Single-walled carbon nanotubes (SWNTs) exhibit high surface areas and precisely defined pores, making them potentially useful materials for gas adsorption and purification. A thorough understanding of the interactions between adsorbates and SWNTs is therefore critical to predicting adsorption isotherms and selectivities. Metallic (M-) and semiconducting (S-) SWNTs have extremely different polarizabilities that might be expected to significantly affect the adsorption energies of molecules. We experimentally and theoretically show that this expectation is contradicted, for both a long chain molecule (n-heptane) and atoms (Ar, Kr, and Xe). Temperature-programmed desorption experiments are combined with van der Waals corrected density functional theory, examining adsorption on interior and exterior sites of the SWNTs. Our calculations show a clear dependence of the adsorption energy on nanotube diameter but not on whether the tubes are conducting or insulating. We find no significant experimental or theoretical difference in adsorption energies for molecules adsorbed on M- and S-SWNTs having the same diameter. Hence, we conclude that the differences in polarizabilities between M- and S-SWNTs have a negligible influence on gas adsorption for spherical molecules as well as for highly anisotropic molecules such as n-heptane. We expect this conclusion to apply to all types of adsorbed molecules where van der Waals interactions govern the molecular interaction with the SWNT.
Authors De-Li Chen, Lynn Mandeltort, Wissam A. Saidi, John T. Yates, Jr., Milton W. Cole, and J. Karl Johnson
The differences in the polarizabilities of metallic (M) and semiconducting (S) single-walled carbon nanotubes (SWNTs) might give rise to differences in adsorption potentials. We show from experiments and van der Waals—corrected density functional theory that the binding energies of Xe adsorbed on M- and S-SWNTs are nearly identical. Temperature programed desorption experiments of Xe on purified M- and S-SWNTs give similar peak temperatures, indicating that desorption kinetics and binding energies are independent of the type of SWNT. Binding energies computed from vdW-corrected density functional theory are in good agreement with experiments.
Authors Joanna Kauczor, Patrick Norman and Wissam A. Saidi
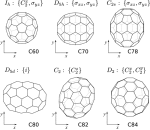
We present frequency-dependent polarizabilities and C 6 dipole-dipole dispersion coefficients for a wide range of fullerene molecules including C60, C70, C78, C80, C82, and C84. The static and dynamic polarizabilities at imaginary frequencies are computed using time-dependent Hartree-Fock, B3LYP, and CAM-B3LYP ab initio methods by employing the complex linear polarization propagator and are subsequently utilized to determine the C 6 coefficients using the Casimir-Polder relation. Overall, the C60 and C70 average static polarizabilities ???(0) agree to better than 2% with linear-response coupled-cluster single double and experimental benchmark results, and the C 6 coefficient of C60 agrees to better than 1% with the best accepted value. B3LYP provides the best agreement with benchmark results with deviations less than 0.1% in ???(0) and C 6. We find that the static polarizabilities and the C 6 coefficients are non-additive, and scale, respectively, as N 1.2 and N 2.2 with the number of carbon atoms in the fullerene molecule. The exponent for C 6 power-dependence on N is much smaller than the value predicted recently based on a classical-metallic spherical-shell approximation of the fullerenes.
Authors W. A. Al-Saidi, Vamsee K. Voora, and Kenneth D. Jordan
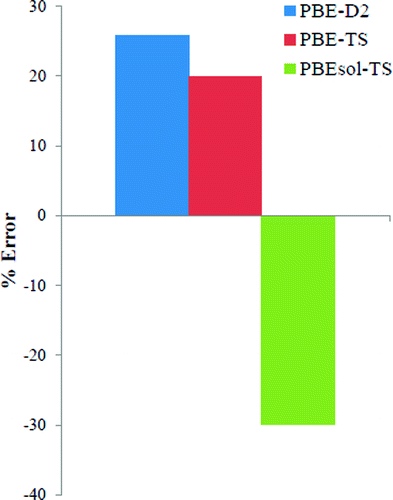
The Tkatchenko–Scheffler vdW-TS method [Phys. Rev. Lett.2009, 102, 073005] has been implemented in a plane-wave DFT code and used to characterize several dispersion-dominated systems, including layered materials, noble-gas solids, and molecular crystals. Full optimizations of the structures, including relaxation of the stresses on the unit cells, were carried out. Internal geometrical parameters, lattice constants, bulk moduli, and cohesive energies are reported and compared to experimental results.